Functional Phospholipid Nano-Microfibers and Nano-Microparticles by Electrohydrodynamic Processing: A Review
Ana C. Mendes and Ioannis S. Chronakis∗
Nano-BioScience Research Group, DTU-Food, Technical University of Denmark, B202, 2800Kgs Lyngby, Denmark
E-mail: anac@food.dtu.dk; ioach@food.dtu.dk
∗ Corresponding Author
Received 13 February 2019; Accepted 06 May 2019; Publication 05 June 2019
Abstract
Functional phospholipid nano-microfibers and nano-microparticles developed using electrohydrodynamic processing methods are reviewed. Depending on the phospholipid concentration the solvent used and the processing conditions, fibers and particles with a range of morphologies, mechanical properties and functionalities were observed. The efficacy of electrospun phospholipid fibers as antioxidant, encapsulation, and delivery matrices for bioactive compounds is also presented.
Keywords: Phospholipids, Electrospray, Electrospinning, Nanomechanics, Drug Delivery.
1 Introduction
Self-assembling molecules such as phospholipids have been substantially investigated due to their excellent properties for applications in industrial products such as pharmaceutics, cosmetics and food additives [1]. In the nature, the cellular membranes are composed of phospholipid bilayers, which is an example of self-assembled structures. The phospholipids arrange themselves by projecting their hydrophobic tails inwards while their polar head groups are exposed on the outside surfaces. The self-assembly of phospholipids in the cell membranes makes them selectively permeable to ions and molecules. Likewise, the unique properties of these molecules allow their self-assembly into other types of different nano structures, including micelles, liposomes, hexagonal, lamellar, cylindrical structures and aggregates, depending on the solvent, the lipid composition and the temperature [2].
Electrohydrodynamic methods, which include electrospinning and electrospray, are cost-effective technologies used to produce high-performance functional nano-micro fibers and nano-microcapsules. The electrospinning technique is a straightforward and versatile top-down manufacturing process for the preparation of functional nano-microfibers. This technique involve the use of a high-voltage electrostatic field to charge the surface of a (bio)polymer or lipid solution droplets, thereby inducing the ejection of a liquid jet through a spinneret. Electrospray differs from electrospinning by the fact that when the polymer concentration is too low, the jet is destabilized due to varicose instability, forming fine highly charged droplets. On the way to the collector the jet/charged droplets (if electrospinning or electrospray, respectively) will partially or fully solidify through solvent evaporation or cooling, forming a dried fiber/particle. A characteristic feature of the electrospinning and electrospray process is the extremely rapid formation of the nano-microstructure (NMS) [3].
The electrohydrodynamics process parameters (applied potential, electric field, spinning distance, flow rate) and fluid parameters (conductivity, viscosity, surface tension, dielectric constant) can be adjusted to tune and optimize the morphologies (e.g. fibers, particles, nano-micro-rods, -tubes, -spirals, -rings) and other characteristics of the nano-microstructures.
In addition, the functionality of the nanostructures produced by electrohydrodynamic process can be achieved through the utilization of blends, coaxial core-shell spinning, inclusion of other functional molecules, and the adsorption of functional components to the surfaces of the nano-microstructures, among other. Polysaccharides (chitosan, alginates, starch, dextrans, xanthan etc.), and proteins (gelatin, casein, zein, egg albumen, etc.) have been electrospun to produce NMS [4, 5].
In this article, the development and the functional properties of electro- spun/electrosprayed phospholipid fibers/particles are reviewed. In particular, the effect of organogel solvents and co-axial electrospinning to tune the diameter and the morphology of the electrospun phospholipid fibers, as well as their mechanical properties characterized by Atomic Force Microscopy are discussed. Furthermore, the efficacy of electrospun phospholipid microfibers as antioxidant encapsulation and delivery matrices for bioactives compounds, are also presented.
2 Phospholipid Nano-Microfibers and Nano-Microparticles by Electrohydrodynamic Processing
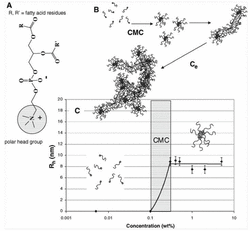
McKee et al., 2006 [6] showed that a phospholipid mixture of asolectin solution above 35% w/w in DMF:CHCl3 [3:2 v/v] is capable of forming continuous fibers using electrospinning processing in normal atmospheric conditions with an average phospholipid fiber diameter around ∼3.3 μm at concentration of 45% w/w. The ability to electrospun phospholipids was explained by changes in the solution rheology based upon the concentration. It was proposed that with the increase of the phospholipid concentration, a progressive transition from single molecules to micelles formation and then to the formation of rod-like structures and creation of elongated cylindrical aggregates occurs, that are responsible to reach a critical length and overlap that allows to be electrospun, as it has been shown for the polymers (Figure 1). They also found that the diameter size (D) did not follow the empirical equation (D[μm] = 0.18(C/Ce)2.7 ) that applies for other polymers based on concentration entanglement (Ce) and solution concentration (C). Accordingly, they found that the micelles were interacting between the formed phospholipid elongated cylindrical aggregates, making the solution highly dependent on viscosity [7]. They confirmed this finding by another self-associating polymer modified with quadruple hydrogen bonding capabilities, and could fit a straight line between diameter and viscosity parameter on a log-log plot. A few years later Hunley et al., 2008[8], showed the ability to electrospin pure 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine in the melted state at temperature around 200º C, due to a rod forming structure at this temperature. The average fiber size was reported as 6.5 ± 2.0 μm and fibers were straight without interconnected points between them.
Moreover, Yu and co-workers [9], electrospun a fibrous network using a mixture of polyvinylpyrrolidone (PVP) with soybean lecithin. This fibrous network upon immersion into an aqueous solutions created liposomes with a very narrow size-distribution and vesicles between 120–370 nm dependent on the parameters applied for electrospinning. Mixtures of water with other organic solvents have been also reported for the electrospinning of synthesized a phosphonium gemini surfactants by Hemp et al., [10]. In particular, the authors tested various spacer link distances, and found that spacers longer than 3 methylenes did not form fibers, while spacerlinks of 2–3 did. Using CHCl3 as solvent, the average diameter of the fibers was about 1.1 μm whereas in H2O:MeOH (1:1 v/v) the diameter was increased to 4–5 μm owing to the increased evaporation point of the solvents.
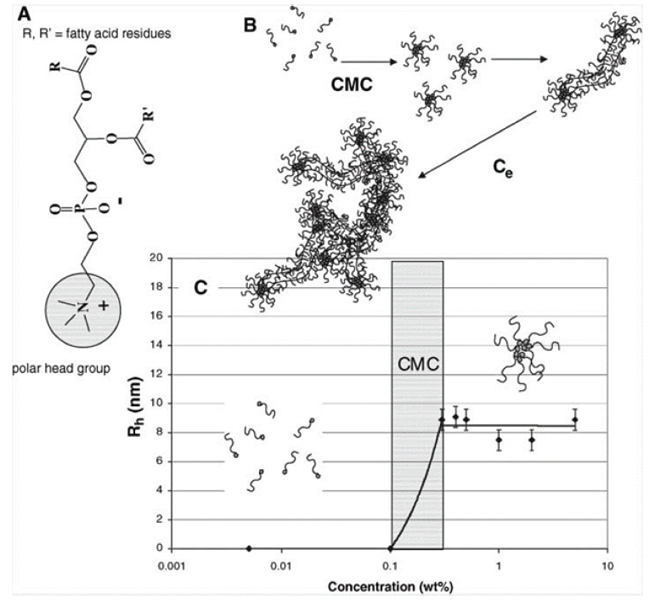
Figure 1 (A) Structure of phosphatidycholine where R and R’ are fatty acid residues with different degrees of unsaturation. (B) Lecithin transition from amphiphilic molecules to entangled, wormlike micelles. (C) Rh values of lecithin micelles in 70/30 CHCl3 /DMF as a function of concentration. The CMC was about 0.1 wt %, and the average spherical micelle was 9 nm [6].
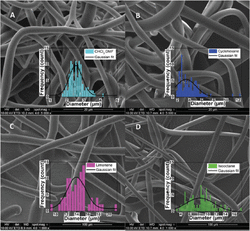
In a recent study from our group, isooctane, cyclohexane and limonene were investigated as solvents to electrospun phospholipid fibers. Those solvents are known to promote elongated self-assembled systems which mimic long-polymers chains as required for electrospinning [11]. The phospholipid concentrations used and the general fiber morphology (beads/particles, fiber and elongated structures) are summarized in Table 1 and the SEM images of asolectin fibers and particles are shown in Figure 2. Electrospun asolectin fibers with average diameters of 2.57 ± 0.59 μm, ∼3–8 μm, ∼4–5 μm and 14.3 ± 2.7 μm were produced using chloroform:dimethylformamide, isooctane, cyclohexane and limonene as solvents, respectively. Overall, it was suggested that the phospholipid concentration and the solvent type dominate the formation of the self-assembled nanostructures, and promote the electrospinning of phospholipid fibers with different morphologies and average diameters. This is in accordance with studies of purified lecithin, which suggested that various solvents promote different changes in the surface curvature of the phospholipids [12], their branching and the aggregation of self-assembled nanostructures, and ultimately the morphology of the electrospun fibers.
Table 1 General morphology of electrospray/electrospun asolectin phospholipids solutions at different solvents [11]
| Phospholipid Concentration [% w/w] | CHCl3:DMF [3:2 v/v] | Cyclohexane | Limonene | Isooctane |
| 30 | Particles | – | – | – |
| 35 | Particles | – | – | – |
| 40 | Elongation | Particles | Particles | – |
| 45 | Fibers | Elongation | Particles | Particles |
| 50 | Fibers | Fibers | Particles | Particles |
| 55 | – | Gelation | Particles | Elongation |
| 60 | – | – | Fibers | Fibers |
| 65 | – | – | Gelation | Gelation |
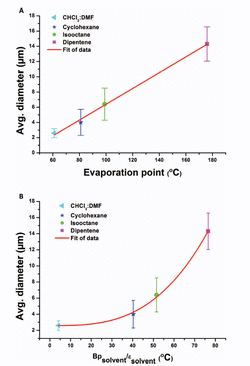
The higher average diameter of electrospun asolectin phospholipid fibers using limonene as solvent in comparison with isooctane is related to the dielectric constant (ɛ solvent) and the evaporation point (Bp solvent) of the solvents. Limonene has a higher evaporation point and dielectric constant (176º C; 2.3) compared to isooctane (99º C, 1.92), thus slower evaporation resulting in fibers with higher average diameter [11].
As shown at Figure 3, the average asolectin fiber diameter (D) can be correlated with both the boiling point and the dielectric constant of the solvent with an allometric function
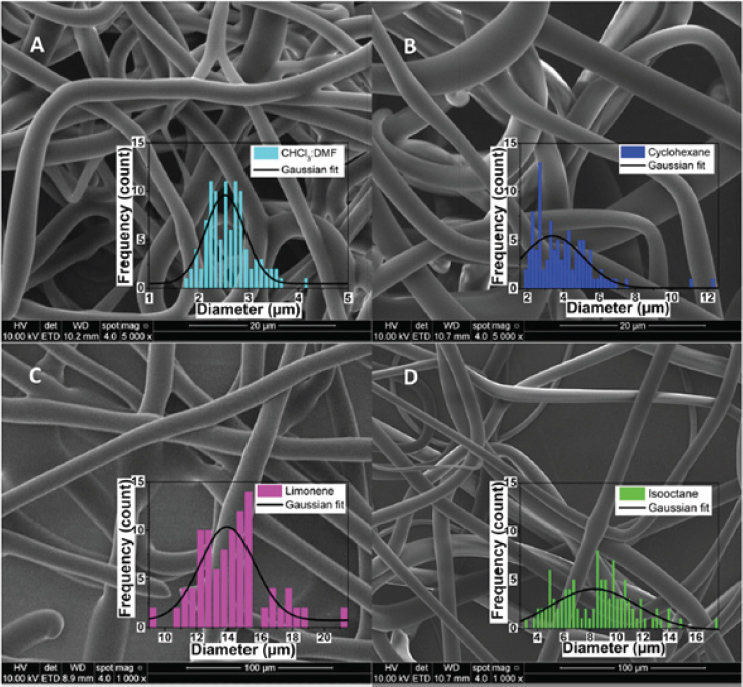
Figure 2 SEM images of electrospun asolectin solution using a single needle: (A) 45% w/w in CHCl3:DMF [3:2 v/v], scale bar 20 μm, (B) 50% w/w in cyclohexane, scale bar 20 μm, (C) 60% w/w in limonene, scale bar 100 μm, (D) 60% w/w in isooctane, scale bar 100 μm. Inserts: histogram of the diameter distribution calculated from 100 random fiber measurements from each sample of electrospun asolectin fibers with different solvents using single-needle electrospinning [11].
The coefficient n is found to be 3.05 ± 0.24 (R2-value of 0.99).
By applying a co-axial electrospinning, where the outer needle contains a pure solvent and asolectin solution in CHCl3 : DMF at the inner needle, a substantial reduction in the fibers diameter was observed due to diminishing of the intermolecular association occurring between the worm-like reverse micelles and reduction of the solution viscosity at the tip of the needles [11].
In particular, the average diameter of the lipid fibers was observed to be reduced to the nanoscale by a factor of 7 (from 2.57 μm to 0.38 μm), when DMF (a solvent with a high dielectric constant) was used as a sheath solvent.
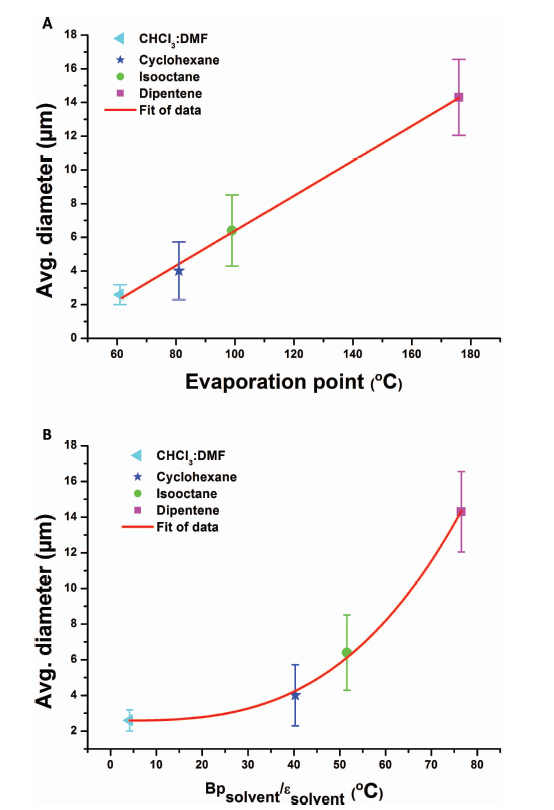
Figure 3 Average diameter of electrospun phospholipid fibers as a function of: (A) solvent evaporation rate and (B) ratio of boiling point and the dielectric constant of the solvent [11].
All the solvents used with the co-axial set-up resulted to a clear lowering of the average fiber diameter compared to the single-needle diameter using the same solution. The lowest average fiber diameter of 0.38 ± 0.14 μm was achieved, using DMF as the outer solvent (Figure 4A–B). This average diameter of 0.38 μm is in close proximity to the theoretical prediction power of the empirical equation (D[μm] = 0.18[C/Ce]2.7 ), that give a predicted value for the electrospun phospholipid fibers of ∼0.35 μm in the absence of intermolecular association between the micelles [11].

Figure 4 SEM images of electrospun 45% w/w asolectin solution in CHCl3 : DMF [3:2 v/v] using co-axial electrospinning. (A) sample Co-A with DMF as the outer solvent, scale bar 5 μm, (B) sample Co-B with CHCl3 as the outer solvent, scale bar 5 μm, (C) sample Co-C, with isooctane as the outer solvent, scale bar 5 μm, (D) sample Co-D, with cyclohexane as the outer solvent, scale bar 30 μm [11].
As discussed above, McKee et al. [6], pointed out that the intermolecular association between the micelles as the main reason behind the increased diameter size. Due to this extensive aggregation, it is most probable that an incomplete split of the jet flow occurs at the single needle set-up. Moreover, it is well known that the dielectric constant of the solvent has a significant impact on the bending instability and the structure of the resulting electrospun fibers [13]. In particular, the accumulation of electric charges passes non-axial instability to the jet flow in the electric field that breaks the jet flow and splits it into thinner ones.
It was suggested that electrospinning of a solution with higher dielectric constant solvents, gives a stronger capability for a jet split in the high electric field (as for example DMF that has a much higher dielectric constant than that of chloroform, isooctane, and cyclohexane [11]. Thus, this might play a pivotal rule in the reduction of the average fiber diameter with the use of the coaxial set-up, due to their significant influence on the jet breakdown properties. Therefore, DMF solvent at the out needle could induce extensive bending instability and enhance Coulombic stretching of the jet, which resulted in extremely finer nanostructures and improved uniformity of the electrospun phospholipids.
It is then reasonable to support that the other solvents (chloroform, isooctane, cyclohexane), although they slightly reduce the average diameter of the fibers using the co-axial process, have dielectric constants that are too low to hold the electric charges and to assist asolectin’s jet in splitting into nanofibers. This is in agreement with the relatively high average fiber diameters, and the wider fiber diameter distribution originated from an incomplete split of the jet flow produced using the above solvents by applying the single needle set-up.
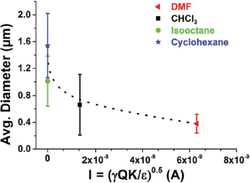
In addition to the dielectric constant, other parameters such as the current intensity, conductivity and the surface tension of the solutions are also crucial for the final diameter of the electrospun fibers [11, 14]. Figure 5, shows the values of the average fiber diameter plotted against the current intensity, I. The results indicate that when the current intensity of the jet was higher, thinner fibers were generated, as the charge density on the surface is known to control the thinning and bending instability during electrospinning. Overall, further studies are needed to investigate the intermolecular association and the aggregation state of the phospholipid micelles at these solvents, the solvent’s evaporation rate at the tip of the needle, and correlate it to the electrospinning processing parameters for the development of phospholipid nano-microfibers with predicted morphologies.
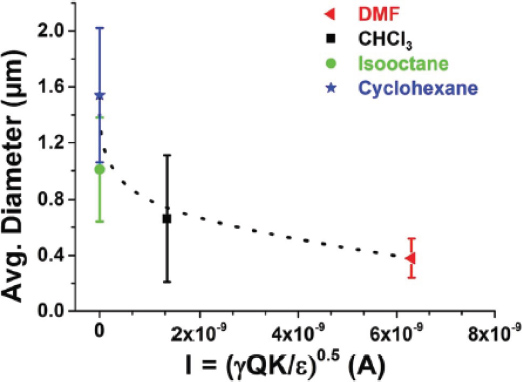
Figure 5 Average diameter of electrospun phospholipid fibers as a function of current intensity (I) using the co-axial set-up (dotted lines are to guide the eye) [11].
Furthermore, by using lower asolectin phospholipid concentration (30% w/w) and electrospray processing, formation of microparticles could be obtained (Figure 6 and Table 1). The obtained partilces were spherical and exhibited a smooth surface with average diameter size of 2.33 ± 1.57 μm that increased slightly to 2.53 ± 1.81 with the encapsulation of 500 μMof capsaicin (unpublished data). Functional hybrid particles could be also formed by electrospraying a mixture of phospholipid with chitosan polysaccharide for the encapsulation of a protein such as ovalbumin (unpublished data).

Figure 6 Electrosprayed phospholipid microparticles produced using a) 30% w/w asolectin dissolved in CHCl3:DMF [3:2 v/v], and b) 30% w/w asolectin dissolved in CHCl3:DMF [3:2 v/v] with 500 μM capsaicin. 20 kV and distance 20 cm (unpublished data).
3 Nanomechanics of Electrospun Phospholipid Fibers
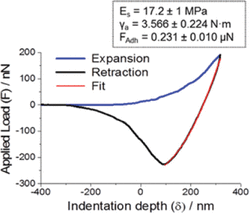
The mechanical properties of electrospun asolectin phospholipid fibers (average diameter of 6.1 ± 2.7μm) were evaluated by nanoindentation using Atomic Force Microscopy (AFM) (Figure 7) [15]. The most remarkable feature in Figure 7 is the high adhesion observed during unloading (black line) and adhesion hysteresis. While adhesion hysteresis is a generally observed phenomenon in loading/unloading cycles by AFM probes on soft matter, Figure 7 is particularly noticeable as the loading process is monotonically repulsive (blue line), a “snap-in” event is absent, and the hysteresis is enormous. It was proposed that a ‘core-surface amphiphilicity’ and the association mechanism of AFM probes with the hydrophilic inner side of the phospholipid fibers, such as hydrogen bonding formation, further contributes to high adhesion hysteresis shown in Figure 7.
The average elastic modulus of asolectin fiber was about 17.2 ± 1 MPa, resembling enhanced elastic properties with respect to the elastic modulus determined for other phospholipid based structures. For instance, the elastic moduli of natural cholesterol and phosphatidylcholine was found to be 0.27 MPa and 0.03 MPa, respectively [16]. Moreover, the elastic moduli of electrospun cholesteryl-succinyl silane (CSS) nanofibers were found to be dependent on the hydrolysis and polymerization degree of CSS molecules [17]. The less polymerized fibers displayed an elastic modulus ranging from 0.8 to 1 MPa, while the elastic modulus of the CSS fibers was determined to be 55.3 ± 27.6 to 70.8 ± 35 MPa [17]. For comparison, the elastic modulus determined for electrospun nanofibers made of synthetic polymers such as poly-L-lactic acid (PLLA), polyvinyl alcohol (PVA) and polyacrylonitrile (PAN) have been found to range from 0.5 to 0.9 GPa [18], 4 to 13 GPa [19] and 5.72 to 26.55 GPa [20], respectively, depending on the size of the diameter of the fiber. In addition, electrospun nanofibers made of natural materials, such as proteins like gelatin, collagen and elastin, exhibited a lower elastic modulus (tensile moduli), compared to synthetic polymers of approximately 426, 262 and 184 MPa, respectively [21].
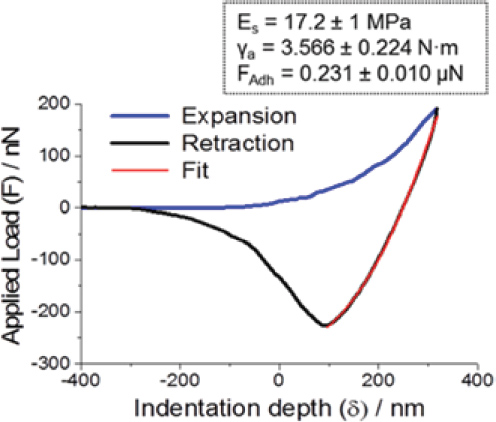
Figure 7 A plot of the Applied Load (F) vs Indentation Depth (δ) for a cycle of piezo expansion-retraction (loading-unloading) of a silicon tip on an asolectin fiber. Inset: Average values of Es, γa and adhesion force FAdh, as obtained from a total of more than 100 force-distance measurements on random locations across five asolectin fibers [15].
Previous studies concerning the determination of Young’s modulus of single electrospun polymeric fibers have found that the Young’s modulus of fibers is regulated by the diameter of the fibers [18,19,22,23] The mechanical behaviour of electrospun fibers was found to be a consequence of the changes in orientation of the polymer molecules during the electrospinning process provided by the strong strain forces of the polymer jets [23]. The high strain rate of the ejected polymer jets induce a molecular orientation of polymer nanofibers along the fiber axis.
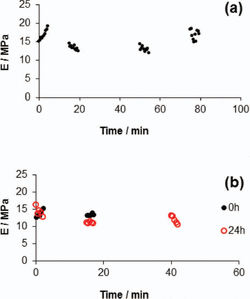
Moreover, the modulus of the fibers was also examined with respect to their stability in ambient conditions [15]. The humidity of the ambient conditions was approximately 60%. As can be seen in Figure 8(a), exposure of the fibers to the humid environment up to 80 min did not affect their nanomechanical properties. In Figure 8(b), another location on the same fiber was also examined, and the modulus remained the same up to 20 min. The fiber was subsequently stored in a desiccator and the same location was examined after 24 h (red circles). The values obtained are statistically indistinguishable from those at 0 h.
Further studies are needed to investigate the effect of solvents and electrospinning processing on the molecular orientation, morphology and mechanical properties of asolectin phospholipid fiber.
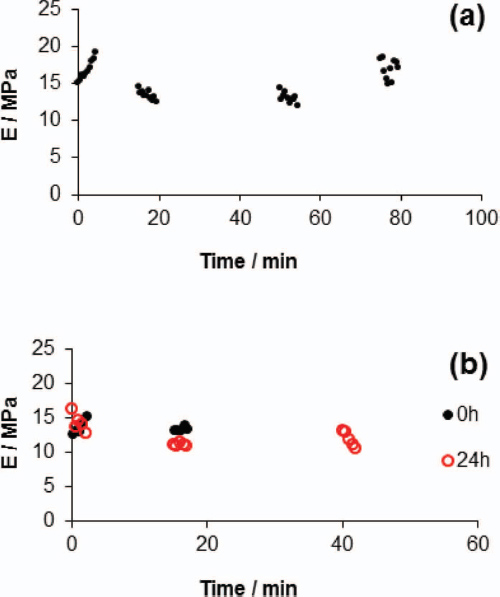
Figure 8 (A) Values of Es at the same location on a fiber as a function of time, obtained under ambient conditions. (B) Values of Es at the same location on a fiber as a function of time, obtained under ambient conditions at 0 h (black dots) and after 24 h of storage in a desiccator (red circles) [15].
4 Functional Properties of Electrospun Phospholipid Fibers
4.1 Phospholipid Fibers as Micro-Encapsulation and Antioxidant Matrices
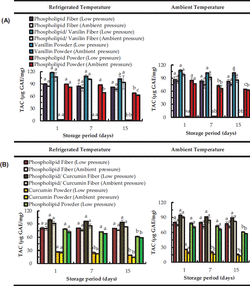
Electrospun phospholipid (asolectin) microfibers were investigated as antioxidants and encapsulation matrices for curcumin and vanillin [24]. These phospholipid microfibers exhibited antioxidant properties which increased after the encapsulation of both curcumin and vanillin. The total antioxidant capacity (TAC) and the total phenolic content (TPC) of curcumin/phospholipid and vanillin/phospholipid microfibers remained stable over time at different temperatures (refrigerated at 4º C, ambient) and pressures (vacuum, ambient) (Figure 9). Particularly, for pure vanillin and curcumin powders (non encapsulated) a decrease in TAC was observed over 15 days of storage.
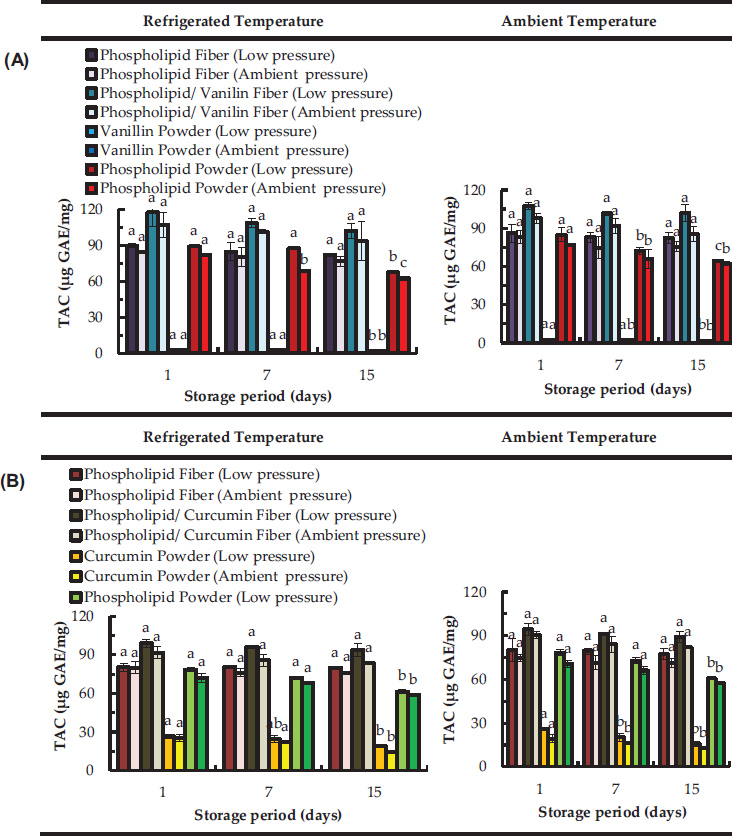
Figure 9 Total antioxidant capacity (TAC) over time of electropsun phospholipid fibers prepared using limonene (top graphs (A) and isooctane (bottom graphs (B)) as solvent; Data are represented as mean ± SD [N = 3]; a–c: significant difference at p ≤ 0.05 in terms of total antioxidant capacity of each storage condition during storage time [24].
The TAC of vanillin stored at 4º C was decreased by 23 (low pressure) and 25% (ambient pressure) from day 1 to day 15. At ambient temperature the TAC of vanillin stored at low pressure and ambient pressure decreased by 30% and 26% respectively from 1 to day 15. After 15 days, curcumin stored at 4º C was observed to decrease significantly its TAC to about 28% and 44% when stored at low pressure and ambient pressure, respectively. At room temperature non-encapsulated curcumin lost around 38 % (at low pressure) and 33 % (at room pressure) of its total antioxidant capacity from day 1 to day 15. Curcumin is a bioactive susceptible to oxidation and its stability is known to be negatively affected by oxygen exposure [25].
However, after encapsulation of phenolic compounds, the TAC of asolectin fibers loaded with both curcumin and vanillin was found to be constant over the time (Figure 9), suggesting that the antioxidant stability of the compounds can be maintained. Moreover, an increase in TAC was observed for fibers loaded with curcumin and vanillin, suggesting an improvement of antioxidant activity due to the combination of phospholipids with phenolic compounds. This is in accordance with previous studies where an improvement of TAC was confirmed by synergistic interactions of lecithins with other phenolic compounds such as tocopherols [26]. The above studies confirm the efficacy of electrospun phospholipid microfibers as encapsulation and antioxidant systems.
4.2 Chitosan/Phospholipid Nanofibers as Delivery Systems and their Cytocompatibility Towards Mammalian Cells
Chitosan (Ch) polysaccharide was mixed with phospholipids (P) to generate electrospun hybrid nanofibers intended to be used as platforms for transdermal drug delivery [27]. Ch/P nanofibers exibithed average diameters ranging from 248 ± 94 nm (Ch/P1) to 600 ± 201 nm (Ch/P3), depending on the amount of phospholipids used. Fibers of Chitosan/Phospholipid at the ration 1:1 (w:w) were designated by Ch/P1, while Ch/P3 is referred for Ch/P at the ratio 1:3 (w:w) respectively. Fourier Transformed Infra-Red (FTIR) spectroscopy and Dynamic Light Scattering (DLS) data suggested the occurrence of electrostatic interactions between amine groups of chitosan with the phospholipid counterparts. Nanofibers were found to be stable at least up to 7 days in PBS solution. The hybrid nanofibers were shown to be stable for at least 7 days in Phosphate Buffer Saline (PBS) solution.
Moreover, cytotoxicity studies (WST-1 and LDH assays) demonstrated that the hybrid nanofibers have suitable biocompatibility [27]. In particular, the cytocompatibility of Ch/P samples was assessed by measuring the cellular metabolism (WST-1 assay) and the plasma membrane integrity (LDH assay). Figure 10a indicates that the metabolic activity of the L929 cells is slightly reduced comparative to the control. However, this reduction was not statistically significant (P>0.05 Student’s t-test). Although the LDH activity levels were significantly increased in cells co-cultivated with nanofibers (Figure 10b), the effect was only marginal especially in comparison to the positive control (Triton X-100).
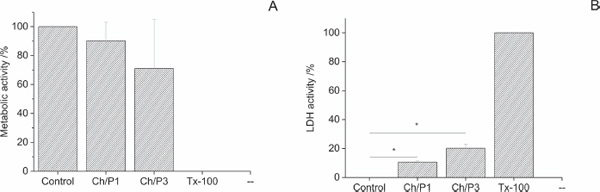
Figure 10 Cytocompatibility measurements of Ch/Phospholipid hybrid nanofibers. (A) Metabolic activity (WST-1 assay) and (B) Plasma membrane integrity (LDH assay) of L929 cells co-cultivated with Ch/P1 and Ch/P3 nanofibers for 24 h. L929 cells seeded in tissue culture polystyrene dishes served as a negative control (control) showing minimal toxicity, while treatment with Triton X-100 (Tx-100) served as a positive control showing maximal toxicity [27].
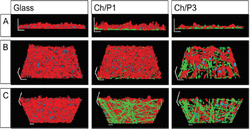
To further analyze the biological compatibility of the nanofibers, the cellular morphology was analyzed by structured illumination fluorescence microscopy (SIFM). F-actin was visualized by Rhodamin-conjugated phalloidin, while chitosan/phospholipid fibers were stained with the recently described chitosan-affinity-protein conjugated to a super folded green fluorescence protein (CAP-sfGFP [28]. As illustrated in Figure 11, cells and nanofibers are in close proximity, suggesting their direct physical interaction. In agreement with the cytotoxicity measurements (Fig 10), the density of the layer formed by L929 cells on top of the fibers was highest on the non-coated glass slides and lowest on the Ch/P3-coated glass. Quantification of the fiber thickness revealed that the diameters of Ch/P1 fibers are thinner than the Ch/P3 fibers. Previous studies have shown that the fiber diameter of electrospun fiber webs influences the cellular metabolic activity [29, 30], and an increase in the fiber diameter, reduced migration, spreading and proliferation.
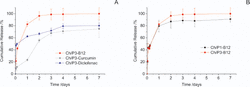
Fluorescence microscopy, also suggested that L929 cells seeded on top of the CH/P hybrid have similar metabolic activity comparatively to the cells seeded on tissue culture plate (control). Finally, the release of curcumin, diclofenac and vitamin B12, as model drugs, from Ch/P hybrid nanofibers was also investigated, demonstrating their potential utilization as a transdermal drug delivery system (Figure 12). The solubility property of the encapsulated compounds control their release profile from the chitosan/phospholipid hybrid nanofibers [27].
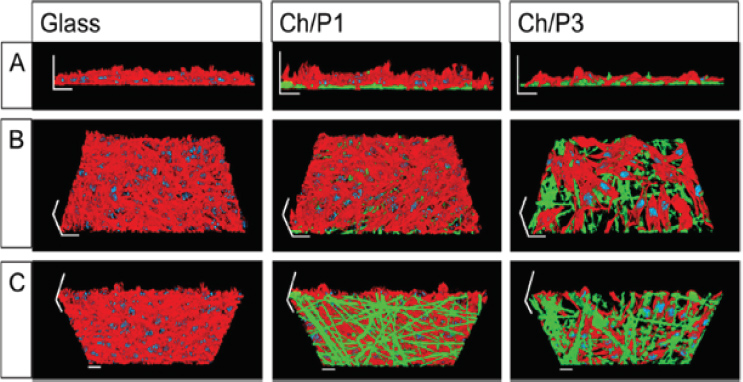
Figure 11 Three-dimensional images of L929 cell layers applying structured illumination fluorescence microscopy. L929 cells were grown on glass, Ch/P1 or Ch/P3 nanofibers. Cellular F-actin was stained with Rhodamin conjugated phalloidin (red). Chitosan fibers were stained with green fluorescence protein conjugated chitosan-affinity-protein (green). Cell nuclei were stained with 40,6-Diamidin-2-phenylindol (blue). (A) front view, (B) top view, (C) bottom view. Scale bars correspond to 20 μm [27].
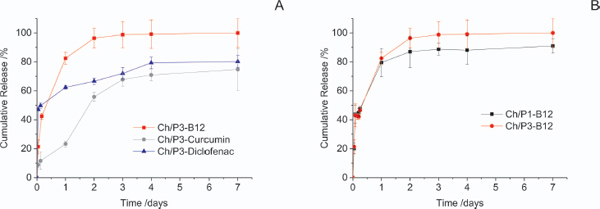
Figure 12 Cumulative release of vitamin B12, Curcumin and Diclofenac from chitosan /phospholipids fibers over 7 days in PBS at 37º C (A). Effect of phospholipid content in the cumulative release of vitamin B12 at the same conditions (B). Each point represents the average value ± standard error [N=3] [27].
4.2.1 Chitosan/phospholipid nanofibers: mechanical and adhesive properties
Furthermore, the effect of phospholipid content on the morphological, mechanical and mucoadhesive properties of the hybrid electrospun chitosan–phospholipid nanofibers was also investigated [31]. Ch/P1 exhibited a stable young modulus of approximately 500 MPa. However, when increasing the content of phospholipid to 1:3 (chitosan: phospholipid), the modulus increased to approximately 750 MPa. Such increase in young modulus was correlated with the increase in the fiber diameter with the increase of the phospholipid content. It could be expected that having a higher content of phospholipids on the formulation would represent more opportunities for molecular rearrangements able to further change the elasticity of the Ch/P fibers. Other studies reported the Young’s modulus of the as-spun crosslinked chitosan fibers and electrospun asolectin fibers to be of 154.9 MPa and 17.3 MPa respectively. Therefore, it is suggested that Ch/P displayed higher elasticity than the pure chitosan or phospholipids electrospun nanofibers, due to the intermolecular bonding between chitosan–phospholipids hybrid nanofibers. Adhesion of the fibers to the hydrophilic silicon AFM probe was also analyzed. The increase in phospholipid decreased the adhesion from 13 to 4 nN. Those trends are in agreement with the mucoadhesion studies of the electrospun Ch/P fibers compressed against sections of small pig intestine. Due to the fact that asolectin phospholipid fibers, which contains lecithin as a major component, shows a fast and /high absorption of water, its mechanical resistance and effectiveness to adhere to the mucus layer is limited when increasing the amount of phospholipids [31].
5 Conclusions
Chloroform:dimethylformamide isooctane, cyclohexane and limonene were proven to be efficient solvents for the production of asolectin fibers and particles with a range of diameters and morphologies depending on the phospholipid concentration and the solvent used. By applying co-axial electrospinning, a substantial reduction in the fibers’ diameter was observed. As shown by using Atomic Force Microscopy the phospholipid fibers were stable in ambient conditions, preserving the modulus of elasticity up to 24 h. At a cycle of piezo expansion-retraction (loading-unloading) of a silicon tip on a fiber, a relatively high adhesion was observed during unloading due to molecular rearrangements at the utmost layers of the fiber caused by the indentation of the hydrophilic tip. Moreover, asolectin phospholipid fibers were observed to have also antioxidant properties that were improved after the encapsulation of the phenolic compounds. The antioxidant capacity of curcumin/phospholipid and vanillin/phospholipid microfibers was observed to remain stable over time at different temperatures (refrigerated, ambient) and pressures (vacuum, ambient), while the pristine non-encapsulated phenolic compounds decreased their TAC and TPC values.
Furthermore, phospholipids/chitosan polysaccharide electrospun hybrid nanofibers can be used as platforms for drug delivery. The nanofibers were shown to be stable for at least 7 days in phosphate buffer saline (PBS) solution due to the electrostatic interactions between amine groups of chitosan with the phospholipid counterparts with controlled mechanical, adhesion properties by the phospholipid content. Overall, further studies are required to fully demonstrate the potential of using phospholipid and phospholipid hybrid fibers particles processed by electrohydrodynamic methods for food, pharma and biomedical applications.
References
[1] J. K. Song, S. H. Yoon, J. S. Rhee and J. J. Han, Chapter 19 Biotechnological Uses of Phospholipids, in Biocatalysis and Biomolecular Engineering, ed C. T. Hou and Jei-Fu Shaw, John Wiley and Sons, Inc, pp. 277–298, 2010.
[2] J. N. Israelachvili, Intermolecular and surface forces, Academic Press, 1991.
[3] A. Frenot and I. S. Chronakis, Curr. Opin. Colloid Interface Sci., 8, 64–75, 2003.
[4] Mendes, A.C.; Stephansen, K.; Chronakis, I.S. Food Hydrocoll. 68, 53–68, 2017.
[5] Mendes, A., Sevilla Moreno, J., Hanif, M., E.L. Douglas, T., Chen, M., and Chronakis, I. International Journal of Molecular Sciences, 19, 2266, 2018.
[6] M. G. McKee, J. M. Layman, M. P. Cashion and T. E. Long, Science, 311(5759), 353–355, 2006.
[7] M. G. McKee, C. L. Elkins and T. E. Long, Polymer, 45, 8705–8715, 2004.
[8] M. T. Hunley, A. S. Karikari, M. G. McKee, B. D. Mather, J. M. Layman, A. R. Fornof and T. E. Long, Macromol. Symp., 270, 1–7, 2008.
[9] D.-G. Yu, C. Branford-White, G. R. Williams, S. W. A. Bligh, K. White, L.-M. Zhu and N. P. Chatterton, Soft Matter, 7, 8239–8247, 2011.
[10] S. T. Hemp, A. G. Hudson, M. H. Allen Jr, S. S. Pole, R. B. Moore and T. E. Long, Soft Matter, 10, 3970–3977, 2014.
[11] L. Jørgensen, K- Qvortrup and I. S. Chronakis RSC Adv., 5, 53644–53652, 2015.
[12] R. Angelico, A. Ceglie, U. Olsson and G. Palazzo, Langmuir, 16, 2124–2132, 2000.
[13] A. Nakano, N. Miki, K. Hishida and A. Hotta, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 86, 011801–011809, 2012.
[14] J. F. Delamora and I. G. Loscertales, J. Fluid Mech., 260, 155–184, 1994.
[15] A. C. Mendes, N. Nikogeorgos, S. Lee, and I S. Chronakis Appl. Phys. Lett. 106, 223108, 2015.
[16] J. M. Crowley, Biophys. J. 13, 711, 1973.
[17] J. Zhang, C. Cohn, W. Qiu, Z. Zha, Z. Dai, and X. Wu, Appl. Phys. Lett. 99, 103702, 2011.
[18] E. P. S. Tan and C. T. Lim, Appl. Phys. Lett. 87, 123106, 2005.
[19] U. Stachewicz, R. J. Bailey, W. Wang, and A. H. Barber, Polymer 53, 5132, 2012.
[20] S. Y. Gu, Q. L. Wu, J. Ren, and G. J. Vancso, Macromol. Rapid Commun. 26, 716, 2005.
[21] M. Li, M. J. Mondrinos, M. R. Gandhi, F. K. Ko, A. S. Weiss, and P. I. Lelkes, Biomaterials 26, 5999, 2005.
[22] E. P. S. Tan and C. T. Lim, Appl. Phys. Lett. 84, 1603, 2004.
[23] M. K. Shin, S. I. Kim, S. J. Kim, S.-K. Kim, H. Lee, and G. M. Spinks, Appl. Phys. Lett. 89, 231929, 2006.
[24] E. Shekarforoush, A.C. Mendes, S. Beeren, I.S. Chronakis, Molecules, 22, 10, 2017.
[25] L.C. Price, Buescher, R.W. Decomposition of turmeric curcuminoids as affected by light, solvent and oxygen. J. Food Biochem. 20, 125–133 (1996).
[26] A. Judde, P. Villeneuve, A. Rossignol-Castera, A. Guillou, J. Am. Oil Chem. Soc. 80, 1209–1215, 2003.
[27] A.C. Mendes, C. Gorzelanny, N.Halter, S.W. Schneider, I.S. Chronakis, Int. J. Pharm. 510, 48–56, 2016.
[28] M. Nampally, B.M. Moerschbacher, S. Kolkenbrock, Appl. Environ. Microbiol. 78, 3114–3119, 2012.
[29] M. Chen, P.K. Patra, S.B. Warner, S. Bhowmick, Tissue Eng. 13, 579–587, 2007.
[30] G.T. Christopherson, H. Song, H.Q. Mao. Biomaterials 30, 556–564, 2009.
[31] A.C. Mendes, J.A. Sevilla Moreno, M. Hanif, T.E.L. Douglas, M. Chen, I.S. Chronakis, International Journal of Molecular Sciences, 19 (8), 2266, 2018.
Biographies

Ana C Mendes holds a licentiate degree in Industrial Chemistry (University of Coimbra, Portugal), a master and a PhD degree from University of Minho (Portugal) in Materials and Biomedical engineering, respectively.
Ana C Mendes conducted her postdoctoral studies in Denmark (Aarhus University and Technical University of Denmark (DTU)). At present she is a senior research scientist at DTU, working within the fields of processing and characterization of food ingredients and biopolymers for applications in food and bioengineering.

Ioannis S. Chronakis received his PhD degree in physical and colloidal chemistry of biomacromolecules from Cranfield University (UK) in 1995. From 1995 to 2001 he was research scientist at Lund University (Sweden) and at INRA, (France). During 2001 to 2010, he was research projects manager at the Swedish Institute for Industrial Research and Development. A faculty member of Technical University of Denmark since 2009, he is actively involved in the development and applications of highly functional electrospun and electrospray structures. His research team is also working in the field of soft nano-bio-structures with tailor-made properties.
Journal of Self-Assembly and Molecular Electronics, Vol. 7 1, 23–44.
doi: 10.13052/jsame2245-4551.7.002
This is an Open Access publication. © 2019 the Author(s). All rights reserved.