Probing the Amyloid Peptide-Membrane
Interaction Using a Liposome
Model System
Liguo Tao1,†, Yunpeng Cao2,†, Cuixia Ma1, Jie Wang2, Lin Lin1,
Lei Liu2,* and Mingdong Dong3
- 1School of Food and Biological Engineering, Jiangsu University, China
- 2Institute for Advanced Materials, Jiangsu University, China
- 3Interdisciplinary Nanoscience Center (iNANO), Aarhus University, 8000 Aarhus C, Denmark
*Corresponding Author: liul@ujs.edu.cn
†These authors contributed equally to this work.
Received 23 March 2016; Accepted 28 July 2016;
Publication 24 August 2016
Abstract
The aggregation of amyloid peptides is closely related to the pathogenesis of degenerative diseases. More and more evidence implies that the proto fibrillar intermediates rather than the mature amyloid fibrils are the toxic species related to the membrane disruption. In this work we found that the self-assembling intermediates of Aβ33-42 during early aggregation are able to break down the liposome. During the process of amyloid peptide (Aβ33-42) intermediate binding to liposome, the β-sheet secondary structure of peptide took change on the molecular level which was characterized by circular dichroism (CD) spectra. The small micelles were formed due to the disruption of amyloid peptide, and further to grow into big irregular complexes with further incubation, which is characterized by the assay of disrupting liposome membrane and atomic force microscopy (AFM). This founding paved the way to understand the interactions between the amyloid peptide and membranes, and support the amyloid peptide nanostructure formed in the early stage of aggregation has good affinity with membrane.
1 Introduction
The so-called amyloid diseases are considered to be closely related to the toxicity of amyloid peptides [1, 2]. Under abnormal physiological circumstances the soluble native amyloid peptides may accumulate in organs or tissues where they form insoluble aggregates, such as fibrils [1–4]. Some of these aggregates are associated with cell impairment [5–8] thatis associated to the pathogenesis of many serious degenerative diseases, such as Alzheimer’s disease (AD) [8–10], type II diabetes [11] and Parkinson’s disease [12, 13], etc. More and more evidence is found that the proto fibrillar intermediates rather than the mature amyloid fibrils are the toxic species [14, 15] and the still soluble amyloid protein nanostructures formed in the early stage of aggregation such as oligomers [5, 16], nanopores [6, 17] and other small amyloid species are considered to be serious damage to the cell [4]. Recent studies of the pathogenesis of amyloid diseases have brought forward increasing evidences indicating the cell membrane as the common target for the intermediates of amyloid peptides. The interaction between the amyloid peptide and the cell membrane results in disruptive structural perturbations, which are closely related to the toxicity of amyloid peptide [18].
Beta amyloid peptide (Aβ) is a typical amyloid peptide derived from the amyloid precursor protein (APP) by proteolysis [9]. The proteolytic fragments are all considered to be related to the pathogenesis of AD disease [9, 10, 19]. For instance, Aβ28 [20], Aβ25-35 [21] and Aβ10-20 [22], form aggregates that were proved to be neurotoxic. Recently Aβ33-42 came into the focus of attention, as it is a significant fragment of the transmembrane domain of APP, so it can target the cell membrane [23, 24] and it is also a key fragment [25] of Aβ42, dominating its aggregation. The structure of relative fragment Aβ34-42 was explored by NMR in previous research. The previous study makes it plausible both to investigate the proto-intermediate aggregation and to find the mechanism of membrane disruption by the intermediates of Aβ33-42. In biological systems, the cell membrane is composed of a complex mixture of lipids and proteins. As we are aiming at fundamental understanding of the interactions between the amyloid peptide and membranes [26], we chose to reduce the complexity by using a liposome as the simplified model system to study the peptide-membrane interaction.
We explored the aggregation behavior of Aβ33-42, initially. The different self-assembled nanostructures, proto-intermediates and mature fibrils, were obtained with different incubation times. It is of most significance that the mechanism of membrane disruption by the intermediates of Aβ33-42 was revealed by the assay of disruption of liposome, atomic force microscopy and circular dichroism spectra (CD). The secondary structure of the amyloid peptide took change from the β-sheet conformation while it adsorbed on the liposomes. This changing could lead to the proto-intermediates disrupting the liposome initially (by the dye release assay) to form small micelles (by AFM).With long-time incubation the micelles aggregated into larger complexes composed of amyloid peptides and lipids. It was proved to be a very suitable model system to understand the interactions between the amyloid peptide and membranes and the disruption of membrane.
2 Experimental Sections
2.1 Material
Aβ33-42 (amino acid sequence: NH2-GLMVGGVVIA-COOH) was purchased from ABBiochem Co. Ltd (Shanghai, China). 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) was purchased from Tokyo Chemical Industry (Tokyo, Japan). Polyvinylpyrrolidone (PVP) was purchased from Aladdin Industrial Corporation (Shanghai, China). Soy Lecithin, Cholesterol, Ethanol, Sodium phosphate dibasic anhydrous, Chloroform and Potassium dihydrogen phosphate were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China).
2.2 Liposome Preparation
Firstly, a mixture of 1 g of Lecithin and 0.2 g cholesterol was dissolved in 20 ml chloroform under ultrasonic conditions and then transferred to a round-bottom glass flask. The solvent was then evaporated in a rotary evaporator (SY-2000, Yarong, Shanghai, China) at room temperature and a thin film of lipid formed on the wall of the flask. The flask was transferred to a Vacuum Drying Oven (DZF-6050, Jing Hong, Shanghai, China) for 12 hours at 60∘C to get rid of the rest of the organic solvent. 0.05 g PVP was dissolved in a phosphate buffer solution with the concentration of 30 mM (pH = 7.2) under sonication for stabilizing the liposome. This buffer solution used to hydrate the lipid film at room temperature (sonication) with the final concentration of the lipid was 10 mM. Multilamellar liposomes (MLV) were formed by vortexing for 4 min in a vortex mixer (Vortex-Genie 2, Scientific Industries, USA) followed by 30 minutes of sonication in a Transsonic Digitals bath sonicator (Elma, Germany), the sonicator was set to 10 seconds working followed by a 5 second break, with a power output of 360 W. The resultant product was centrifuged at 6,000 rpm for 15 minutes. The supernatant was extruded through a filter with 0.22 μm pore size to obtain the final liposome.
2.3 The Preparation of Aβ33-42 Aggregation Intermediates Solution
1mg Aβ33-42 powder was dissolved in 1ml 1,1,1,3,3,3-Hexafluoro-2-propanol, with 5 seconds ultra-sonication followed by 5 seconds of vortex mixing. Sonication and vortexing were repeated 6 times. The solution was put in a thermo-shaker (PHMT, Grant Instruments, UK) for 8 hours at 350 rpm at 25∘C, to ensure that peptides powder had been fully dissolved. 100 μl of Aβ33-42 solution was transferred into a 1.5 ml centrifuge tube, and sealed with parafilm. The tube was put in a Vacuum Drying Oven for 1 hour at 25∘C. After that, the HFIP was fully evaporated and a thin film of peptide formed on the bottom of tube. 200 μl Milli-Q water was added to the dessicated-film tube, the solution was sonicated for 5 seconds and vortexed for 5 seconds (repeated 6 times) until the solution became clear. Finally, the solution was incubated for 300 min on a thermo-shaker at 350 rpm and 37∘C. The Aβ33-42 peptides aggregated into intermediates with a final concentration of 500 μM after 1 hour incubation.
2.4 Sample Preparation for Atomic Force Microscopy, Dye Release Assay, Circular Dichroism Studies
250 μL prepared liposome solution was diluted with 250 μL Milli-Q water in a 1.5 mL centrifuge tube and 300 μL of peptide intermediate intermediates solution was added. This was mixed on a thermo-shaker for 6 hours at 350 rpm and 37∘C. Sets of samples, for Atomic Force Microscopy, circular dichroism spectroscopy and dynamic light scattering characterization were taken out from the mixture after 1 hour, 3 hours and 6 hours, respectively.
2.5 Turbidity Measurements
The solution of Aβ33-42 was incubated for 300 min on a thermo-shaker at 350 rpm and 37∘C. Set of samples were taken out from the solution for the turbidity measurement. Turbidity was measured at room temperature on a fluorescence spectrophotometer (PerkinElmer LS55) using 1 cm path length quartz cell. Both excitation and emission wavelengths were set to 400 nm with spectrum band width of 1 nm. The signal was quantified by averaging the emission intensity at 400 nm (slit width = 2.5 nm) over 15 s in an attenuate mode.
2.6 Atomic Force Microscopy Analysis
10 μL of the complexes (liposome and intermediates, with a concentration of intermediates 500 μM) were deposited on freshly cleaved mica for 10 min, after that the residue liquid on the surface was removed. The sample was rinsed 3 times with Milli-Q water and dried in air before the measurement. All AFM measurements were performed in air using a commercial Asylum Research MFP-3D-SA (Asylum Research, Santa Barbara, California, USA) in tapping mode with a silicon cantilevers (OMCL-AC160TS-R3, Olympus). Cantilevers had a nominal spring constant of 26 N/m and a normal tip radius of 15 nm. The resonant frequency of the cantilever is 300 kHz in all measurements and a scan frequency was set at 1 Hz with optimized feedback parameters. The resolution of all the original AFM images was 512 × 512 pixels per image.
2.7 Secondary Structural Changes Measured by Circular Dichroism Spectroscopy
The fresh solution of Aβ33-42, intermediate and fibril, as well as the complex of Aβ33-42 and liposome is characterized by Circular Dichroism (CD). CD measurements were performed on a Spectropolarimeter (JASCO-J1500,). The spectral region from 190 nm to 250 nm was scanned with a scan speed of 100 nm/min, with a path length of 0.1 cm and a slit-width of 2 nm. All experiments were carried out at 25∘C in a 1-mm quartz cuvette. For each sample, the baseline, the signal of Milli-Q water, was subtracted from all measurement results. The sample volume for CD measurement 350 μL. The concentration of peptide intermediates and the one in the liposome complex was both 250 μM.
2.8 Dye Release Assay
Liposomes were prepared as before, the lipids were then re-suspended in 12 mL of PBS (20 mM, pH = 7.3) containing 1 mg/ml calcein sodium salt. The mixture was centrifuged at 6,000 r/min for 15 minutes, and the supernatant was extruded through a 0.22 μm filter. 1.3 mL of this solution was centrifuged at 15,000 r/min for 30 minutes to separate the excess calcein in the solution. The supernatant was removed, and the sediment was rinsed with 0.2 ml PBS buffer. The release of calcein from calcein-containing vesicles was measured by a Hitachi F-4500 fluorescence spectrophotometer at room temperature with excitation at 496 nm and emission at 515 nm. Data were collected over 2 hours at 5 second intervals.
3 Results and Discussion
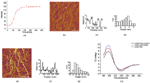
We initially investigated the aggregation of Aβ33-42 in bulk solution (Milli-Q water) by turbidity measurement (Figure 1a). It clearly showed the typical sigmoidal growing curve of amyloid peptide aggregation. We obtained the intermediates and mature amyloid aggregates by incubating Aβ33-42 for different time. Figure 1b and 1e show the AFM images of the intermediates and the fibrils. The intermediates which had been incubated for 1 hour at 37∘C were determined to be the amyloid films with the height ∼4 nm (Figure 1c and 1d.). Figure 1e shows the AFM image of fibrils which have been incubated on the thermo-shaker for 6 hours at 37∘C. The height of the mature fibrils assembled from Aβ33-42 was measured to be ∼9 nm (Figure 1f), and the histogram distribution was presented in Figure 1g. The morphology and height distribution of the intermediates and mature fibrils assembled from amyloid peptides show a clear difference. Furthermore, the secondary structure of Aβ33-42 was characterized in peptide fresh solution, intermediate and fibril by CD. We found that dominating secondary structure of Aβ33-42 aggregates was β-sheet at all times from early aggregation to mature fibrils (Figure 1h). We suggest that the β-sheet is the secondary structure that dominates its polymerization. Based on previous research [5, 6], the amyloid intermediates from early aggregation could be more likely to interact with the phospholipid bilayer of cell membrane. Therefore, it is plausible to investigate the interaction of liposome (mimic membrane) and amyloid intermediates.
Before investigating the interaction between the amyloid peptide and liposome, the liposome was prepared by optimal protocols obtained after several trials with different surfactants or different ratios of components with fixed surfactant. The prescription of agents was composed of egg L-a-phosphatidylcholine(PC) (20 mg/ml, 1000 mg) and cholesterol (C) (4 mg/ml, 200 mg), chloroform (20 ml), phosphate buffered saline (PBS) (50 mL) and polyvinylpyrrolidone PVP (50 mg) which is necessary as well in the liposome formation. The size of prepared liposomes was characterized by AFM. Figure 2a–c display the AFM images of liposome incubated for (a) 1 hour, (b) 3 hours and (c) 6 hours. Figure 2d–f showed the corresponding height distributions. Fitting the histograms with a Gaussian estimates the average height to be 73.5 ± 11.8 nm, 72.4 ± 13.0 nm and 72.5 ± 12.2 nm. AFM results suggest that the pure liposome is stable without apparent changes in the morphology and height obtained on mica surface. Although it is not the original liposome structure as dried one on mica in ambient condition, it could be repeatable with no changing in the liposome environment without others. It could be considered as the reference when the liposome was disrupted, and the dried liposome structure should be different.
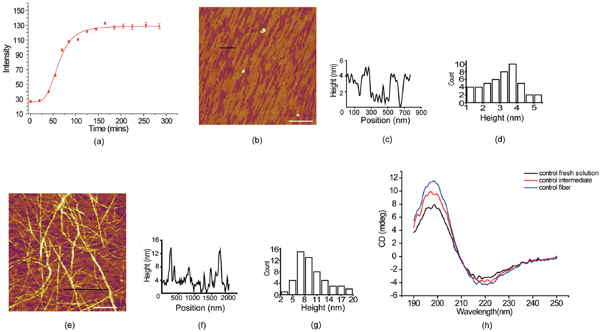
Figure 1 The aggregation behavior of Aβ33-42 by turbidity measurement and AFM images of the intermediates and the fibrils assembled from Aβ33-42. (a) Aggregation of Aβ33-42 in bulk solution characterized by turbidity measurement. (b) Intermediates of Aβ33-42 incubating 1 hour. (c) Height profile along the black line in (b). (d) The histogram distribution of the height of the intermediates. (e) Fibrils of Aβ33-42 incubating 12 hour. (f) Height profile along the black line in (e). (g) The height histogram distribution of amyloid fibrils. The scale bar was 2 μm. (h) CD spectra of amyloid peptide fresh solution, intermediates and fibrils.
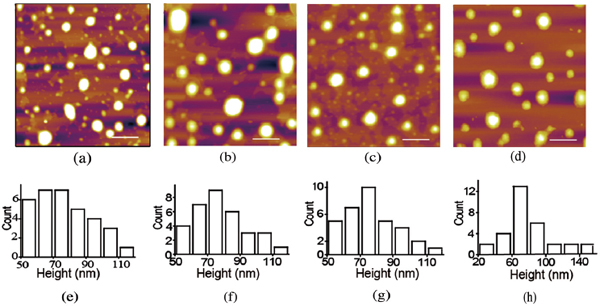
Figure 2 The height distribution of liposome with different incubation time characterized by AFM. (a–d) AFM images of liposome after different incubation time. (a) 0 hour, (b) 1 hours, (c) 3 hours, (d) 6 hour. (e–h) The histogram of the height distribution of liposomes after different incubation time. The scale bar was 2 μm.
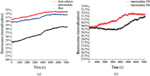
To investigate the interaction mechanism between the liposome and amyloid intermediates, we started with the membrane disruption assay induced by Aβ33-42 assembly. Figure 3a presents the calcein dye release efficiency of liposomes disrupted by Aβ33-42 fresh solution, intermediates and fibrils, at a concentration of 500 μM. It is not hard to observe that Aβ33-42 assembly has the ability to disrupt the liposome although it is not strong (Control assay of Triton-X100 and buffer solution is in SI). The intermediates assembled from Aβ33-42 display the good ability to disrupt the liposome compared to the peptide in fresh solution and fibril. However, it should mentioned that amyloid fibril showed the similar disruption ability after 7,000 min in the dye release assay, which means that some possible interaction mechanism of amyloid peptide rupturing the membrane might exist. However, the intermediates of amyloid peptide seem the dominating species in the disruption of liposome, especially in the beginning, the intermediates presents obvious effect on the liposome disruption compared to the amyloid fibril. We also investigated the dye release efficiency of Aβ33-42 intermediates at different concentration (Figure 3b), and unsurprisingly the higher concentration of Aβ33-42 intermediates disrupted more liposomes.
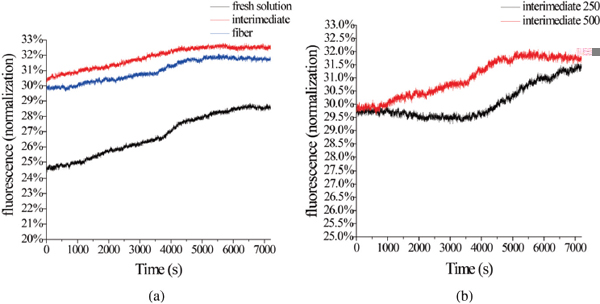
Figure 3 Disruption of liposome membrane induced by Aβ33-42 (a) The calcein dye release of liposome trigged by Aβ33-42 in fresh solution, intermediate and fiber at the concentration of 500 μM over the same time. (b) The calcein dye release of liposome trigged by Aβ33-42 intermediates at the concentration of 250 μM and 500 μM.
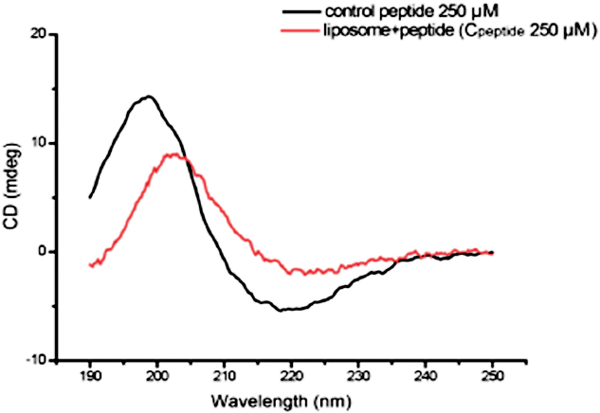
Figure 4 The secondary structure of amyloid peptide intermediates characterized by CD spectra.
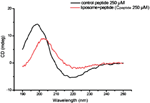
In addition, we tested whether the secondary structure of the amyloid peptide changes when Aβ33-42 interacts with liposome. Figure 4a shows the CD-spectra of pure amyloid intermediates and complexes of liposome and intermediate. As there is obviously observable shift in the peak position of β-sheet secondary structure of amyloid peptide (Figure 4), we conclude that the secondary structure of peptide changes when the peptide intermediates bind to liposomes. It is suggested that liposome could tune the peptide secondary structure when the interaction occurred between them. Finally, it could result in the liposome disruption or some complex formation.
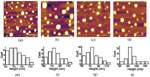
It is therefore plausible that we noted drastic changes in the morphology of the dried liposome on mica in the presence of amyloid peptides. The liposome structure could be affected due to the amyloid peptide intermediate disruption. Figure 5 reflected the interaction between the liposome and the intermediate of amyloid peptide from AFM observations. The morphologies of the complex of liposome and intermediate are displayed in Figure 5a–c with different incubating time. It shows that the morphology of the complex of liposome and amyloid intermediate with 1 hour incubation differs from the pure liposome. The size of the complex was much smaller than the pure liposome. The height of the complex (Figure 5a) was measured to be 27.1 ±10.5 nm (Figure 5d) compared to the liposomes with the height of 73 nm characterized by AFM. When the amyloid intermediates were incubated with the liposome for 3 hours, the complex grew into bigger ones (Figure 5b). The height of the complex was determined to be 49.8 ±13.6 nm and 90.6 ±12.5 nm presented in Figure 5e. After the incubation of liposome with amyloid intermediates for 6 hours, some irregular structures (Figure 5c) with more than twice size of normal liposome occurred. The height of these irregular structures was measured to be 78.8 ± 11.2 nm and 152.2 ± 10.2 nm (Figure 5f). We could deduce that the amyloid intermediate disrupted the liposome from these morphology changing and size variation of the complex of amyloid intermediate and liposome. The summary and the comparison of the size of liposomes both in the absence of amyloid intermediates and in the presence of amyloid intermediates were presented in Figure 5g. The clear difference was displayed. The longer of interaction time of amyloid intermediates with liposome, the more extent of liposome interruption presents. Amyloid fibril did not show this effect.
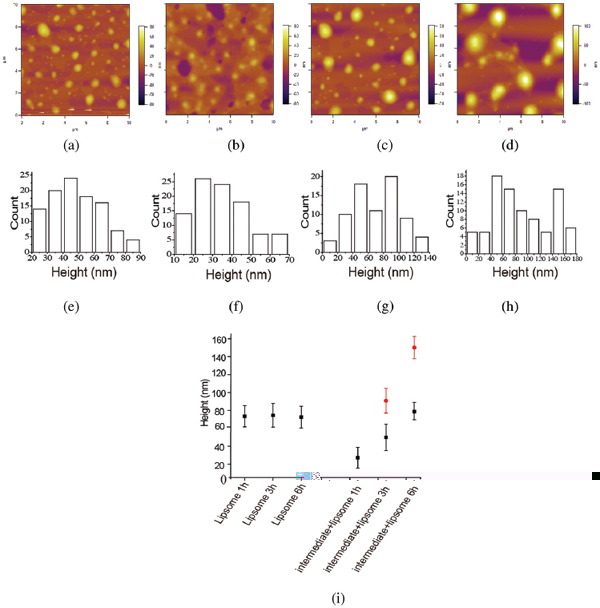
Figure 5 AFM images and the height histogram distribution of the complex of liposome and amyloid intermediate, incubated in different time: (a) 0 hour, (b) 1 hour, (c) 3 hours, (d) 6 hours. The height distribution of the complex, incubated in different time: (e) 1 hour, (f) 3 hours (g) 6 hours. (h) The height scattergram of liposome and the complex of liposome and amyloid intermediate obtained from AFM results. A lot of liposomes have been measured to determine the height, and the histogram is obtained from the height measurement of liposome done by the software nanoscope analysis.
In this work, we have already explored the interaction between the liposome and amyloid intermediates. The disruption of liposome by the intermediates was revealed clearly. During the process of the interacting between the amyloid peptide and liposome, the secondary structure of Aβ33-42 changed and the content of β-sheet decreased. The secondary structure was affected after the liposome mix with amyloid peptide. It is very likely to be the interface effect of liposome on amyloid peptide secondary structure. The interaction between amyloid peptide and liposome occurred on the interface of liposome, amyloid peptide adsorbs on the liposome initially, and some binding interaction happened there. The interface of liposome will be very likely to mediate the secondary structure variation of amyloid peptide. The beta strand of pristine peptide will be affected, which is observed from CD spectra in the manuscript. It is implied that the mechanism of peptide interrupting the liposome, is not only the structure of liposome changed, but the secondary structure of amyloid peptide was also affected, and it happened simultaneously. After that, the complex could be resulted from the interaction between peptide and liposome and it was proved from the observation in Figure 5. The possible mechanism that the amyloid intermediate affected the liposome was proposed. The liposome is made of phospholipid bilayer. Amyloid peptide and the intermediates can break the liposome structure. Aβ33-42 was originated from the transmembrane domain of the cell, and it is therefore plausible that the peptides and the intermediates would like to bind to the phospholipid bilayer. With the gathering of intermediates molecules on the liposome, the liposome will be attacked more, which might result in the formation of the complex of phospholipid and amyloid peptide (micelles). It was proved by AFM in Figure 5a. After that, theses micelles can grow up into the big complex and more micelles and some amyloid peptide and intermediates merged together. Finally, the irregular complex was formed that was proved by AFM in Figure 5c. To sum up, the mechanism of the amyloid peptide interrupting liposome was that peptide disrupted the liposome to form small micelles with different sizes, and these micelles and amyloid intermediates jointed together to become bigger particles. The mechanism of amyloid peptide interrupting the liposome is very likely to pave the way on understanding the possible mechanism responsible for the cell membrane interruption of amyloid peptide aggregates.
4 Conclusion
To sum up, amyloid intermediates assembled from Aβ33-42 were obtained with controlled incubation time. The most importance is that the mechanism of membrane disruption by the intermediates of Aβ33-42 was revealed by dye release assay, atomic force microscopy and circular dichroism spectra. On the molecular level, the β-sheet secondary structure of amyloid peptide changed during the process of interacting of peptide with the liposomes. Furthermore, the intermediates of amyloid peptide could disrupt the liposome initially to form small micelles, after that these micelles composed of amyloid peptides and lipids further merged into larger aggregates on the nanoscale. The results obtained in this work provided a possible mechanism responsible for membrane disruption and further shed light on understanding the interaction between the amyloid peptide and membranes.
Conflict of Interest: The authors declare no competing financial interest.
5 Acknowledgements
This work was supported by Youth Natural Science Foundation of Jiangsu Province (Grant number: BK20140528, BK20140013) and the Jiangsu University research foundation (Grant number: 14JDG021). This work was also supported by Jiangsu Planned Projects for Postdoctoral Research Funds (1401068B) and Jiangsu University Funds (14JDG061). Financial support from Institute for Advanced Materials, Jiangsu University is also gratefully acknowledged.
References
[1] G. G. Glenner, D. Ein, E. D. Eanes, H. A. Bladen, W. Terry and D. L. Page. Science, 174, 712–714 (1971).
[2] G. G. Glenner, N. Engl. J. Med. 302, 1333–1343 (1980).
[3] G. G. Glenner and C. W. Wong. Biochem. Biophys. Res. Commun., 120, 885–890 (1984).
[4] D. J. Selkoe. Nature, 426, 900–904 (2003).
[5] M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft and W. L. Klein. Proc. Nat. Acad. Sci. U.S.A., 95, 6448–6453 (1998).
[6] H. A. Lashuel, D. Hartley, B. M. Petre, T. Walz and P. T. Lansbury. Nature, 418, 291–291 (2002).
[7] M. Fandrich, M. A. Fletcher and C. M. Dobson. Nature, 410, 165–166 (2001).
[8] C. M. Dobson. Philos. Trans. R. Soc. Lond. B Biol. Sci., 356, 133–145 (2001).
[9] E. D. Roberson and L. Mucke. Science, 314, 781–784 (2006).
[10] M. Goedert and M. G. Spillantini. Science, 314, 777–781 (2006).
[11] A. Lorenzo, B. Razzaboni, G. C. Weir and B. A. Yankner. Nature, 368, 756–760 (1994).
[12] W. Dauer and S. Przedborski. Neuron, 39, 889–909 (2003).
[13] T. M. Dawson and V. L. Dawson. Science, 302, 819–822 (2003).
[14] R. Kayed, Y. Sokolov, B. Edmonds, T. M. McIntire, S. C. Milton, J. E. Hall and C. G. Glabe. J. Biol. Chem., 279, 46363–46366 (2004).
[15] Y. Sokolov, J. A. Kozak, R. Kayed, A. Chanturiya, C. Glabe and J. E. Hall. J. Gen. Physiol., 128, 637–647 (2006).
[16] W. L. Klein. Neurochem. Int., 41, 345–352 (2002).
[17] A. Quist, L. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso and R. Lal. Proc. Nat. Acad. Sci. U.S.A., 102, 10427–10432 (2005).
[18] S. M. Butterfield and H. A. Lashuel. Angewand. Int. Edn., 49, 5628–5654 (2010).
[19] D. L. Brody, S. Magnoni, K. E. Schwetye, M. L. Spinner, T. J. Esparza, N. Stocchetti, G. J. Zipfel and D. M. Holtzman. Science, 321, 1221–1224 (2008).
[20] E. Mikros, D. Benaki, E. Humpfer, M. Spraul, S. Loukas, C. I. Stassinopoulou and M. Pelecanou. Angewand. Int. Edn., 40, 3603–+ (2001).
[21] M. S. R. Shearman, C. I. Ragan and L. L. Iversen. Proc. Natl. Acad. Sci. U.S.A. 91, 1470–1474 (1994).
[22] L. Liu, L. Zhang, L. Niu, M. Xu, X. B. Mao, Y. L. Yang and C. Wang. AcsNano, 5, 6001–6007 (2011).
[23] J. Herms, C. Priller, T. Bauer, G. Mitteregger, B. Krebs and H. A. Kretzschmar. J. Neurosci., 26, 7212–7221 (2006).
[24] D. J. Selkoe, Trends Neurosci., 16, 403–409 (1993).
[25] L. Liu, L. Zhang, X. B. Mao, L. Niu, Y. L. Yang and C. Wang. Nano Lett., 9, 4066–4072 (2009).
[26] L. Liu, L. Niu, M. Xu, Q. S. Han, H. Y. Duan, M. D. Dong, F. Besenbacher, C. Wang and Y. L. Yang. AcsNano, 8, 9503–9510 (2014).
Biographies

L. Tao obtained the bachelor degree in biological sciences at Anhui Normal University (Anhui, China). Liguo Tao is now a mater student of School of Food and Biological Engineering in Jiangsu University (JiangSu, China), The research interest focuses on the mechanism and antimicrobial activity of antimicrobial peptides.

Y. Cao obtained the bachelor degree in Huaiyin Institute of Technology (JiangSu, China). Yunpeng Cao is a master student of School of Materials Science and Engineering in JiangSu University (JiangSu, China), and the research project is studying on the interaction between amyloid peptide and the liposome.

C. Ma was born in 1990, and she graduate from Yantai University in 2014 majoring in Marine biological resources. At present, she is studying Master of food quality and safety in Jiangsu University.

J. Wang graduated from Chinese Academy of Science. Since 2013, he became the assistant research in Institute for Advanced Materials of Jiangsu University. His work concentrates on the interaction of nanomaterials and proteins, especially of disease-related biomarker, structure and function of complex. Current research focuses on the interactions of nanomaterials and amyloid protein.

L. Lin was born in 1978. He received his Ph.D. degree from Jiangsu University. His Ph.D. research was focused on Food Science. Since 2008, he has been working at the Jiangsu University as associate research fellow in the School of Food and Biological Engineering. he continue his postdoc study at Interdisciplinary Nanoscience Center, Aarhus University (Denmark). His research is now focused on antimicrobial preservation of natural products, new food packaging materials, and the use of oils and fats active ingredients.

L. Liu has completed his Ph.D in 2010 from National Center for Nanoscience and Technology, China, after that he went to iNANO Center of Aarhus University, Denmark for postdoctoral studies about 3 years working with Prof. Flemming Besenbacher and Dr. Mingdong Dong. He became the Young Distinguish Professor of Jiangsu University China, in 2013. Since 2014, he has been the research and Jiangsu Specially-Appointed Professor of China. His research interest focuses on the scanning probe microscopy, molecular self-assembly of peptides and etc.

M. Dong achieved Ph.D. in 2006 in Department of Physics and Astronomy, then he continue his postdoc study at Rowland Institute, Harvard Harvard University. Since 2009, he worked as an associate professor in Bio-SPM group, Interdisciplinary Nanoscience Center, Aarhus University (Denmark). Mingdong Dong long-term commits to the development and application of scanning probe microscopy techniques. He successfully expanded biomolecular imaging application in a liquid phase with high resolution. He developed for the first time the use of force microscopy microsecond. His interests focus on the structure of the material and research functions.
Journal of Self-Assembly and Molecular Electronics, Vol. 1, 1–18.
doi: 10.13052/jsame2245-4551.2016001
© 2016 River Publishers. All rights reserved.