Computational Strategies for Protein-Surface and Protein-Nanoparticle Interactions
Received 17 October 2014; Accepted 1 December 2014
Publication 31 March 2015
Journal of Self-Assembly and Molecular Electronics, Vol. 2, 1–26.
doi: 10.13052/jsame2245-4551.211
© 2015 River Publishers. All rights reserved.
Giorgia Brancolini*, Laura Zanetti Polzi and Stefano Corni
- Center S3, CNR Institute Nanoscience, Via Campi 213/A, 41125 Modena, Italy
- *Corresponding Author: giorgia.brancolini@nano.cnr.it
Abstract
Protein-nanoparticle associations have important applications in nanoscience and nanotechnology but the recognition mechanisms and the determinants of specificity are still poorly understood at the microscopic level. Crucial questions remain open, related to the association mechanisms, control of binding events, and preservation of functionality. Gold is a promising material in nanoparticles for nanobiotechnology applications because of the ease of its functionalization and its tunable optical properties. We present a concise overview of recent computational modeling advances which were pursued in the quest for a theoretical framework elucidating the association mechanisms and the ability to design and control the recognition events of a specific class of systems, namely, interfaces between polypeptides/proteins and a gold surface in the presence of water. We select two different methodological advances, the first related to the effect of surfactants covering the surface of nanoparticles and altering their interactions with proteins and the second related to the immobilization of proteins on inorganic surfaces and conserving their functionality. Both cases, demonstrate how the understanding of the polypeptide-surface coupling mechanisms is essential to the control of the process and exploitation for biotechnological and nanotechnological purposes.
Keywords
- gold
- nanoparticles
- amyloid
- docking
- molecular dynamics
- redox potential
1 Introduction
The interaction between proteins and solid surfaces is of fundamental importance for a variety of scientific and technological problems, ranging from biomedicine to nanoelectronics. Combinatorial biotechniques have shown that the molecular recognition capability of proteins can be specifically oriented toward inorganic surfaces, opening-up the way for new concepts in self-assembling. [1] The interaction of proteins with nanostructured surfaces or nanoparticles is also the key to several biological processes occurring in vitro and in vivo. [2] However, a detailed understanding of such interactions is lacking.
Experiments in this field are extremely challenging since they need to probe the interface between the biomolecule and the solid interface in realistic aqueous environment. Some relevant issues that remain open from the experimental work are e.g. the difficulty of quantifying the affinity and orientation of proteins to surfaces, the alteration of the protein structure at surfaces and the role of the environment. [3–5]
Computer simulations have emerged as a powerful tool for treating the structural and functional properties of complex assemblies which can effectively complement experimental studies of protein-NP and protein-surface systems. [6–11] In the last few years, the use of recently developed simulation methodologies and current generation force fields had permitted novel applications of molecular dynamics simulation, which have enhanced our understanding of different physical processes governing peptide-surface binding [9, 11–23] and amino acid driven synthesis of NPs. [24] However, there are several challenges that must be tackled when treating these systems in computations. The first difficulty is related to the several length scales spanned by these systems going from the atomic (local interactions between amino acids and the surface) to the nm of even μm one, when considering the formation of layers of bound proteins on the surfaces or around the nanoparticles (i.e. protein corona). A second fundamental problem to be addressed is the need for an extensive sampling of conformations which is mandatory for gaining insights into the binding process but also for a proper description of the functional properties of the dynamical complexes (see Section 2.3.3). In fact, a proper sampling of the conformational space is not only needed to properly account for entropic contributions to binding, but also to account for the different protein conformations that may exist in solution, and interact with the surface. Therefore, the development of multiscale approaches to treat hybrid protein-inorganic systems is a fundamental computational target. MD simulations are the single-scale method that presently better compromises between accuracy and size of simulated systems and time length of the simulations. However, MD simulations covering the huge diversity of protein-surface systems are often lacking of accurate and consistent force fields allowing the description of real inorganic systems whose complexity may be related e.g. to the variety of interaction sites (faces, edges, vertexes) or to the presence of surfactants covering the surface and altering the interaction with water and proteins as will be discussed in Section “The Role of Citrate: the Force Field”. Moreover, proteins and solid surfaces or nanoparticles have been traditionally studied by different scientific communities, and the computational tools developed for one kind of systems (e.g., proteins) are not often compatible with the tools developed for the others.
All these situations represent yet unexplored variables in the field of protein-inorganic surface and nanoparticle interactions, that require further extension of computational tools (eg., development of proper force fields) and methodologies (e.g. calculation of the reduction potential for bioinorganic interfaces).
In the last few years, a classical atomistic force field to describe the interaction of proteins with gold (111) surfaces was developed and applied in our group and in collaboration with others. [25–28] Based on the novel parameters developed, various levels of computational description of protein-surface interactions were implemented and applied involving classical Molecular Dynamics and Brownian dynamics, that cover multiple length- and time-scales.
In this paper we focus on two theoretical challenges in the field of modeling protein-surface interactions and on progress made to meet these challenges. We select two different methodological advances, the first related to derivation of force field parameters for the description of citrate surfactants covering the surface of nanoparticles in solution, in a consistent and compatible way with the existing GolP [29] for the protein-AuNP surface interactions (Section 2.2); the second related to the extension of the perturbed matrix method (PMM) [30] to the calculation of the redox potential of a covalently bound protein to a gold surface (Section 2.3). In both cases, the results demonstrate how the understanding of the peptide-surface coupling mechanisms is essential to the control of the process and exploitation for medical and nanotechnological purposes.
The paper is organized as follows. The two methodological advances are presented separately and in a sequential form. In Section 2.2 we present our proposed computational strategy combining Brownian Dynamics (BD) rigid-body docking [31] and classical molecular dynamics (MD), [29] used to study the molecular driving forces guiding the binding of a given protein to gold NPs (GNPs). More in detail in Section “The Role of Citrate: the Force Field” we describe how we incorporated the stabilizing adsorption layer of GNPs surfactants (citrate in this case) through the development of a suitable force-fields (FFs) able to describe the citrate anion and their interfacial physisorption on the GNP. In the same section we report the preliminary results on the nature of the binding of a fibrillogenic protein β2-microglobulin to the citrate-coated gold surface.
In Section 2.3 we describe the second methodological advance based on the extension of the QM/MM methodology to treat redox proteins on solid surfaces. [32] Also for this computational strategy, in Section “Theory” we introduce the theoretical background and the issue to be tackled. In Section “Gaining Insights into the Reduction Potential Variations” we review the results relative to a prototypical protein system on a gold electrode to understand the redox thermodynamics of proteins covalently bound to a gold surface.
In Section “Conclusions” we discuss how computational strategies may help the understanding of the polypeptide-surface coupling mechanisms and potentially allow the control of the assembly process and its exploitation for biotechnological and nanotechnological purposes.
2 β2-Microglobulin on Citrate-Coated Au(111)
2.1 The Strategy
In order to study protein-inorganic surface association processes, we have implemented a multi computational strategy whose pipeline includes the following steps:
- rigid body docking with Brownian Dynamics (BD)
- docked configuration clustering
- atomistic MD refinement (MD)
The pipeline makes use of different software packages implemented in a consistent and compatible way at the different scales such as the SDA software [33, 34] for the rigid body docking using the ProMetCS continuum solvent model for protein-gold surface interactions [31] and the GROMACS package [35] for MD simulations using the GolP [29] force field.
The main purpose of the first step of the procedure is to perform an extensive sampling of three-dimensional space in an efficient way and to generate reasonable configurations of protein-surface encounter complexes by following the diffusional association of the protein towards the surface. In protein-surface docking with SDA, the protein is modeled as a particle diffusing in a solvent that is treated as a continuum exerting frictional and random, stochastic forces on the protein; while the metal surface is described as a large multi-layers cluster placed in the XY-plane. More specifically, the Au(111) surface is described by a very large gold cluster with three atomic layers. During the calculations, the position of the metal is fixed with the centers of the atoms in the surface layer at z = 0, i.e., in the xy plane of the simulation box. The surface area of the metal has to be larger than the size of the adsorbate in order to account for interatomic interaction effects up to the cutoff employed in calculations. The adsorption of proteins to metal surfaces are described with a continuum solvent in BD, accounting for specific properties of the hydration shell on metal surfaces by including additional, semi-empirically parameterized terms in the protein-surface forces. [31] The starting position and orientation of the protein is generated randomly at a given distance from the surface, which defines the limit where the protein-surface interaction energy becomes negligible. At each BD step a protein-surface interaction energy and the force acting on the protein are computed using the implicit-solvent ProMetCS force field, developed and parameterized for protein-gold surface interaction. [31] The ProMetCS energy includes the following terms: (i) a sum of pair-wise interatomic Lennard-Jones terms that describes van der Waals and weak chemical interactions between the biomolecule and the gold surface (based on the fully atomistic force field GolP); [29] (ii) the electrostatic interaction free energy in the water environment, which includes an image-charge interaction energy of the protein effective charges with a flat infinite, overall uncharged, metal surface along with the direct Coulomb interaction of protein effective charges with the charges placed at each Au atom, mimicking a gold surface non-zero potential. Electrostatic energy also includes a desolvation correction term which takes into account the change of the electrostatic interaction energy due to polarization and distortion of the hydration shell at small protein – Au surface distance; (iii) a protein hydrophobic desolvation and (iv) a surface desolvation free energy term arising from the partial replacement of the metal hydration shell by the protein. All the energy terms are computed with a procedure which is mostly grid-based and is therefore efficient for application to long-time simulations of protein binding processes.
The main purpose of the second step of the procedure is to cluster the stored docking configurations so that only a small number of representative structures need to be used for the atomistic MD refinement. Millions of putative protein-docking complexes can be generated in a few hours on a commodity cluster by this BD. The low-energy protein-surface complexes resulting from the docking are therefore clustered, by means of a hierarchical clustering algorithm, according to their conformational similarity to reduce the number of structures. The number of clusters that represents the data appropriately varies in different docking cases. We use an empirically derived threshold to determine how many clusters represents the ensamble of docked configurations appropriately. This threshold is defined as the distance between clusters which is in turn defined as the average backbone RMSD between docked protein structures in one cluster relative to structures in another cluster. The representatives of the most populated clusters from the second step are chosen and used in subsequent third step of the multi computational procedure.
The main purpose of the third step of the procedure is to refine the selected rigid body docked orientations of the protein respect to the surface with a more detailed model of the system including full conformational flexibility, to obtain plausible structures of the bound complex. This step is accomplished with the use of fully atomistic MD simulations assessing the stability of the docked encounter complexes and treating protein structural relaxation. The extensive sampling of the conformational space is needed to account for the different protein conformations that may exist in solution, and interact with the surface.
2.2 The Role of Citrate: The Force Field
The surface of GNPs can be covered with surfactants that alter their interaction with water and proteins. Citrate is one of the most common GNP surfactants, being used during nanoparticle synthesis to prevent aggregation. Since citrate does not bind strongly to gold, it might be fully or partially replaced by the protein upon binding. In this section, we illustrate the theoretical challenge made to incorporate the role of citrate surfactants in the protein-surface binding within the computational strategy described in the previous section.
The OPLS/AA force field that we adopted in the simulations does not contain parameters for the citrate anions which are interacting with the gold surface and that we wish to simulate. Therefore, we generated force field parameters for the citrate-coated GNPs consistent with the OPLS/AA force field. To this aim, we performed structural optimization and electronic structure calculations starting from the single molecule structure of the citrate anion at DFT level of theory and localized basis set, B3LYP/6-31G* using the Gaussian09 computer packages. The missing bond and angle parameters were generated from a Hessian matrix determined at the B3LYP/6-31G* level of theory, according to [36]. Charges were derived by fitting the ab initio ESP grid points following the Merz-Kollman scheme, [37] and from these a set of RESP (Restrained ElectroStatic Potential) atomic charges was derived. The interactions of the GNP with the surfactant are including the dispersive forces, which are described with a Lennard-Jones potential. To model these interactions, the atom-atom 12-6 LJ parameters were obtained using the mixing rule which were shown to reproduce the experimental interaction energies for a test set of molecules, for more details see [29].
To our knowledge, the formation of a citrate ad-layer composed of interacting citrate molecules as a stabilizing layer has never been incorporated in simulations due to the lack of suitable force-fields (FFs) able to describe the citrate anion [38] and their interfacial adsorption on the top of the GNP. Moreover, the structural details of citrate anions adsorbed on the GNP surface are not clear in experiments [39] since the charge state of the gold surface is unknown. Citrate covered gold surfaces are certainly negative but the underlying layer of gold cores are supposed to be positively charged according to electrochemistry experiments [40].
As a detailed molecular mechanics model for cit-GNPs (12, 14), we propose a surface in which the fully deprotonated citrate anions are described as interacting adsorbed species on a positively charged GNPs. The deprotonated state of the citrate anion was derived assigning the standard protonation state at pH 6.8 with H++, [41] corresponding to the physiological pH. Among the various crystal surfaces, Au(111) is the most stable and therefore the most commonly occurring in nanoparticle preparations [42]. For this reason we consider an Au(111) face of the NP in our model. The regular citrate ad-layer on the top of Au(111) was generated with a ratio of the surface gold ion and citrate concentrations suitable to reproduce experimental electrochemical data on the cit-GNPs [43, 40].
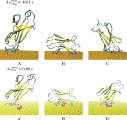
We first examined the stability of the citrate ad-layer on the top of Au(111) in aqueos solution by using 20 ns of standard MD simulations at 300 K. The initial and final distribution of citrate anion on the positively charged GNP is shown in Figure 1. We remark that none of the citrate was displaced from the surface during the entire lenght of the simulation in explicit water. The initial ordered distribution of citrate was stabilized on the top of the GNP surface by direct contact with the surface gold atoms. No large distortion of the ad-layer from the initial conformation have been observed.
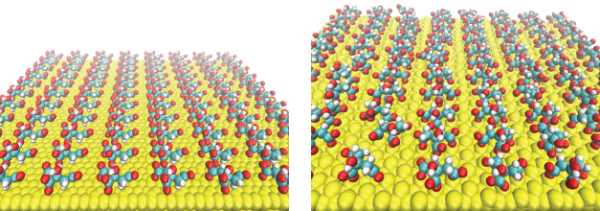
Figure 1 Initial (on the left) and final (on the right) citrate anion distribution on positively charged Au(111) after 20 ns of classical MD with GolP and OPLS/AA in SPCE water.
2.3 The Surface-Protein Binding
In this article, we present a preliminary study of the proposed molecular mechanics model for citrate-coated GNPs interacting with β2m, one of the best characterized amyloidogenic protein with medical relevance.
To investigate the nature of the binding between β2m and a citrate-coated gold surface, as well as the effect of a negative surface potential, we looked for the possible adsorption orientations of β2m on citAu(111) (and the corresponding driving forces) by initially using the protein-surface docking method implemented in SDA 6.0 [44], and we then studied the stability of the docking orientation by MD simulations. More in detail, we introduced: (i) an implicit electrostatic effects of negatively charged citrate molecules on the surface assigning a small negative charge density per surface atom and (ii) an explicit citrate molecule on a neutral Au surface incorporating the steric hindrance and the novel GolP parameters in the ProMetCS force field [31]. The direct interaction of the protein with an explicit layer of citrate molecule will be investigated later on in the article.
During docking, the interaction energy of the protein with the Au(111) surface is given by three main terms: [31] van der Waals energy described by site-site Lennard-Jones, ELJ, interactions, adsorbate-metal electrostatic interaction energy, UEP and the desolvation energy of the protein, , and of the metal surface, (values are reported in Table 1). As already mentioned, the electrostatic term arises from surface polarization and is represented by an image-charge term [45].
Table 1 Resultant encounter complexes from rigid-body BD docking of β2m (1JNJ) to an Au (111) surface. A hierarchical clustering algorithm (based on a minimum distance linkage function) was applied to the diffusional encounter complexes after docking to a bare negative gold surface. The reported complexes represent 98 per cent of the encounter complexes and 92 per cent obtained by BD simulation, respectively.
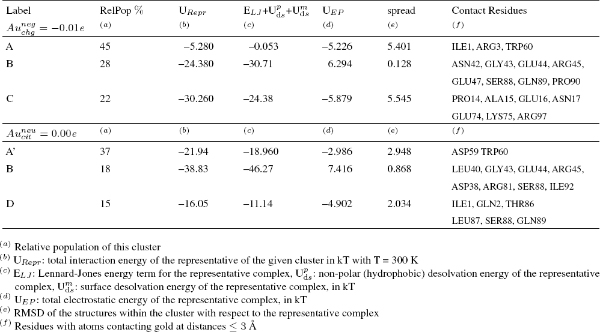
Table 1 Resultant encounter complexes from rigid-body BD docking of βm (1JNJ) to an Au (111) surface. A hierarchical clustering algorithm (based on a minimum distance linkage function) was applied to the diffusional encounter complexes after docking to a bare negative gold ( and -0.01 e) surface. The reported complexes represent 98 per cent of the encounter complexes and 92 per cent obtained by BD simulation, respectively.
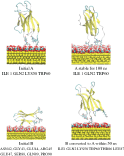
When this docking procedure was applied to the system, it yielded three different orientations accounting for more than 92 per cent of the total encounter complexes when the negative Au surface was considered and it yielded three different orientations accounting for more than 70 per cent of the encounter complexes, when the explicit citrate molecule was instead used (see Table 1). The representative structure in both cases of each computed complex is shown in Figure 2. The protein residues contacting the surface differ in the various complexes, and are listed in Table 1.
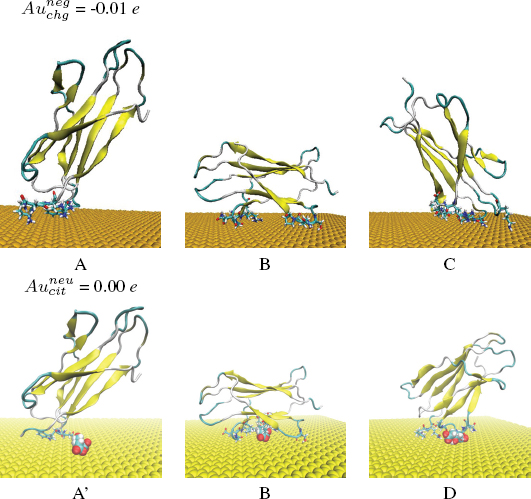
Figure 2 Most populated encounter complexes of β2m on negatively charged gold nanocluster obtained by BD simulation. (a) In the case of , the structures of representative complexes for each of the three clusters are shown, ordered by decreasing cluster size. The reported complexes represent the 92 per cent obtained by BD simulation. (b) In the case of , the structures of representative complexes for each of the three clusters are shown, ordered by decreasing cluster size. The reported complexes represent the 70 per cent obtained by BD simulation. The protein backbone is shown in cartoon representation. The residues contacting the gold surface are shown in stick representation. The citrate anion in Van der Waals representation.
(i) In the case of the negatively charged state of the gold surface the preferred orientation are complex A which is still involving the residues at the N-terminal (ILE1, ARG3) tail and DE-loop (LYS58, ASP59 and TRP60) of the protein but complexes B (driven mostly by ELJ interactions) and C (driven both by ELJ and electrostatic terms) are also present. (ii) In the case of docking in the presence of a negative citrate molecule bound to a neutral surface the ranking becomes A > B > D which gives a consistent picture with respect to the proposed model also in the presence of the steric hindrance associated to the citrate molecule on the surface. In both cases poses A and B are present, therefore suggesting a picture in which the layer of citrate covering the surface of the GNP does remain on the surface of the gold and interacts with the protein, as already predicted by the proposed atomistic model in Figure 1. We remark the presence of a different complex D, in which β2m binds the surface via the N-terminal tail (ILE1, GLN2) and Loop FG (THR86, LEU87, SER88, GLN89) not stable in the presence an extended negatively charged coverage of surface (due to citrate anions) because it is probably based on the fortuitously favorable contact of some residues with the single citrate.
The stability of the present encounter complexes resulting from the rigid docking was assessed performing classical MD simulations. We started with the most representative and populated complexes obtained from rigid-body BD docking (complexes A, B) and we first used 50 ns of standard MD simulations at 300 K in explicit water and ions. In the case of the most stable complex A the simulation was extended to 100 ns. All simulations were based on the GolP [29] force field with the SPC/E water model as implemented in the GROMACS package [35].
In the case of complex A, the protein remained stable with the same global orientation with respect to the surface of cit-Au(111) during the entire lenght of the simulation which was extended up to 100 ns. In the case of complex B, the protein rapidly reoriented to a new orientation equivalent to complex A with contact patches involving ILE1, ARG3, HIS31, ASP59 and TRP60, as shown in Figure 3 in agreement with the docking prediction thus strenghtening the role of the electrostatic interactions on the binding orientation. Analysis of the conformational changes of the protein data requires more extensive sampling methods and will be the subject of a forthcoming paper as well a direct comparison with experimental results. [46]
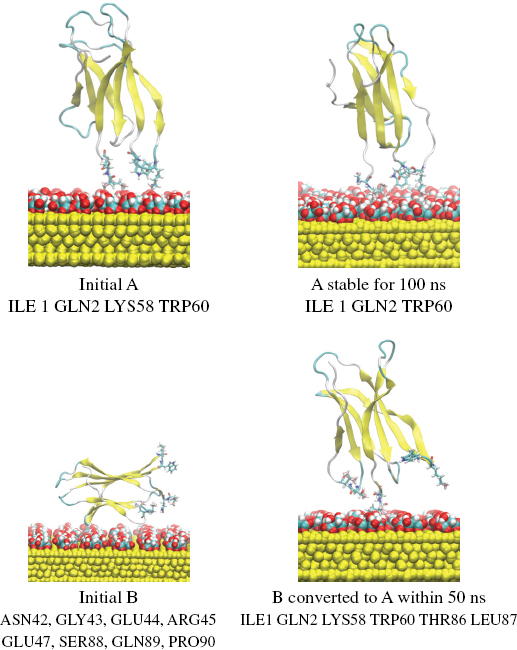
Figure 3 Orientations of β2m (1JNJ) on the cit-Au(111) surface obtained following the MD refinement. Simulations of different lenght were performed on the representative of two most stable and populated assemblies A and B. The initial and final global orientation of the systems after the simulation are reported.
3 The Case of Cytochrome C Adsorbed on Au(111)
3.1 The QM/MM Methodology for Redox Proteins on Solid Surfaces
In the present section, we illustrate the use of a hybrid quantum mechanics/ molecular mechanics (QM/MM) methodology to treat of redox proteins adsorbed on solid surfaces. The Perturbed Matrix Method (PMM) [30] has been recently applied to describe several chemical processes in complex molecular systems [47–49] and has been recently extended to treat the bio-inorganic interface. [32] In the first subsection, the PMM is briefly outlined and in the second subsection a specific application of the methodology to a protein adsorbed on a solid surface is presented.
3.2 Theory
In PMM calculations [30, 50–53], similarly to other QM/MM procedures [54–56], a portion of the system to be treated at electronic level is pre-defined (the quantum center, QC), with the rest of the system described at a classical atomistic level exerting an electrostatic perturbation on the QC electronic states. However, in the PMM, instead of including directly in the Hamiltonian operator the perturbation term, as usual in other QM/MM approaches, the effect of the perturbation is obtained by diagonalizing the perturbed Hamiltonian matrix constructed in the basis set of the unperturbed Hamiltonian eigenstates. The essence is thus to use high-quality unperturbed electronic states as a basis set to express the Hamiltonian matrix of the quantum center including the electric field perturbation resulting from the atomic environment. With a relatively low computational cost, the PMM can be therefore applied to a very large set of molecular configurations, hence providing the dynamical coupling of electronic properties with classical degrees of freedom. Indeed, the phase space sampling is provided by classical molecular dynamics (MD) and thus a statistically relevant sampling of the quantum-center/environment configurations can be achieved, which is necessary for a proper description of functional properties in dynamical, complex systems. The method proved to be able to provide a reliable modeling of different chemical processes, preserving both the quantum-chemical description of the chemical event and the configurational complexity of the overall atomic-molecular environment. [47–49] The general PMM scheme is given here.
- The first step is to pre-define the portion of the system responsible for the chemical event (in the case of charge transfer processes, for example, the electron donor/acceptor sites) to be treated at the electronic (quantum) level. The rest of the system is, instead, described at a classical-atomistic level exerting an electrostatic perturbation on the quantum-center electronic states.
- The second step involves the calculation of an orthonormal set of unperturbed electronic Hamiltonian eigenfunctions on the isolated quantum-center by means of ab initio or DFT calculations on the equilibrium structure in vacuum.
- Fully classical MD simulations of the whole system (quantum-center and external-environment) are carried on in order to extensively sample the phase space of the system.
- In the final step, for each configuration of the whole system generated from the MD simulation the perturbing effect of the external environment on the quantum-center eigenstates is included. This is achieved by constructing and diagonalizing the perturbed electronic Hamiltonian matrix constructed in the basis set of the unperturbed Hamiltonian eigenstates of the quantum center. Indicating with and E the perturbing electric potential and field, respectively, exerted by the environment on the QC:
where and are the QC total charge, dipole operator and unperturbed electronic eigenfunctions, respectively, ΔV approximates all the higher order terms as a simple short range potential, is the identity matrix and the angled brackets indicate integration over the electronic coordinates. The diagonalization of provides a set of eigenvectors and eigenvalues representing the QC perturbed electronic eigenstates and energies. When only the perturbed electronic ground-state energy shift is needed for the calculation of the property of interest with no atomic positional changes involved, ΔV can be disregarded.
For the more specific task of calculating the reduction potential of redox-active proteins, the perturbed electronic ground-state energies are calculated for the QC reduced and oxidized chemical states, providing the perturbed electronic ground-state energy shift. Also, the perturbing environment configurations are obtained by classical MD simulations performed in both the reduced and oxidized ensemble (i.e., with the QC in the reduced and oxidized chemical state, respectively).
The (Helmholtz) free energy change associated to the electron transfer reaction (related to the reduction potential via , with F the Faraday constant) is calculated using the following equation [57]:
In the above equation is the whole system energy change upon reduction (oxidation), , with and representing the perturbed ground-state electronic energy of the red (reduced) and ox (oxidized) chemical states, respectively, and use was made of the approximation , i.e., the environment internal energy change associated to the QC reduction is disregarded being exactly zero within the present description. [53] and are the perturbed electronic ground-state energies evaluated at each MD frame via the PMM approach and averaging is performed in either the oxidized or reduced ensemble as indicated by the subscript of the angle brackets.
Although Equation 3 is based on an exact relation in principle, given the sampling problems of finite-time simulations, the best estimate of the reduction free energy is obtained by averaging the values provided by the reduced and oxidized ensembles: [58]
3.3 Gaining Insights into the Reduction Potential Variations
The previously outlined methodology was recently applied to study the redox thermodynamics of iso-1-cytochrome c (Cyt c) covalently bound to a gold surface. Cyt c is a widely investigated redox protein of well known biochemical relevance and it served as a prototypical system to understand the thermodynamics and kinetics of redox proteins since a long time. [59] Also, it is a suitable candidate for bioelectronic applications as it is able to carry out fast and reversible electron transfer with a bare gold surface with retention of protein functionality. [60] Recently, the PMM was applied to calculate the reduction potential of Cyt c adsorbed on gold, comparing the results to previous experimental ones. [60, 61] The results are discussed in detail elsewhere [32] and are here only briefly summarized.
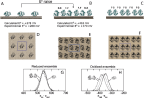
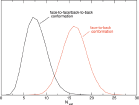
As previous electrochemical measurements on Cyt c pointed to a relevant role of the concentration of the proteins on the electrode surface in the determination of the redox potential, [60, 61] the PMM procedure was applied to two sets of classical MD simulations, mimicking different coverages of the electrode surface. In the first set, one protein bound to a gold surface is considered in a periodic box while in the second set two adsorbed Cyt c inside a periodic box are considered, reproducing respectively a low and high concentration of proteins on the surface (see Figure 4, panels A to F). The QC of the system, necessary for the PMM/MD procedure to be applied (see previous subsection), includes the atoms of the prosthetic group and the side chains of the axial ligands (in the system containing two proteins, only one of these groups is treated at the quantum level). Long MD simulations of the two previously described systems were performed (100 ns for the single Cyt c system and 180 ns for the system containing two proteins) in both the reduced and oxidized ensemble, providing an extensive sampling of the perturbing environment, composed by the rest of the protein(s), the solvent, the gold surface and the counterions used to neutralize the periodic box. In addition, supplementary shorter MD simulations and subsequent PMM calculations were performed for the high concentration system in order to test an alternative orientation between the two proteins in the periodic box. Indeed, three limit reciprocal orientations are possible for the two proteins: calling face the side of the protein in which the heme group is exposed and back the opposite side, these three orientations are the face-to-face, the back-to-back and the face-to-back. In the system used for the main calculations both the face-to-face and the back-to-back orientations are present thanks to the use of the periodic boundary conditions. The face-to-back conformation was tested separately.
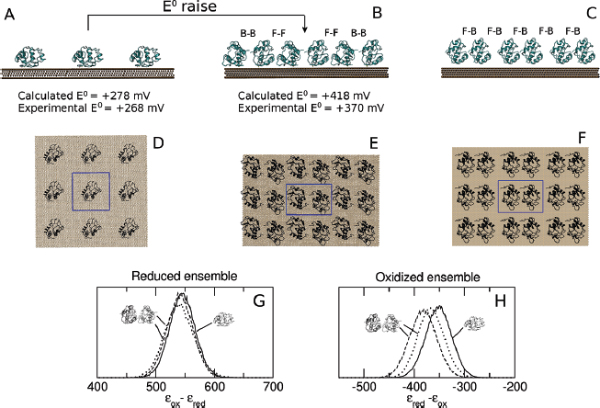
Figure 4 Side view (panels A, B and C) and top view (panels D, E and F) of the low (A and D) and high (B and E for the face-to-face/back-to-back conformation and C and F for the face-to-back conformation) concentration systems. The central box (highlighted in panels D, E and F with blue lines) is surrounded by its periodic images on the electrode plane. For the sake of clarity, water is not shown. In panels B and C the reciprocal orientations between the two proteins in the two high concentration systems analyzed is also highlighted: face-to-face (F-F), back-o-back (B-B) or face-to-back (F-B). Panels G and H: normalized distribution of the perturbed ground-state energy difference for the low (continuous line) and high (dashed line for the face-to-face/back-to-back conformation and dotted line for the face-to-back conformation) concentration systems in the reduced (G) and oxidized (H) ensembles. Energy values in kJ/mol.
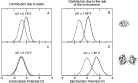
As previously mentioned, experimental electrochemical measurements highlighted the importance of the concentration of the proteins on the electrode in the determination of the redox potential: in particular, between the low and high concentration case a difference of +102 mV in the reduction potential is observed. [61, 60] Such a difference is very well reproduced by the calculated results orientation, that provide a difference between the corresponding E0 values of +140 mV (see Figure 4). In Figure 4, panels G and H, the distribution of the perturbed ground state energy difference is reported for the two simulated systems, showing that the overall shift of the redox potential in the high concentration system is due to a shift of the energy difference in the oxidized ensemble, i.e. to a destabilization of the oxidized state in the high concentration system. Such a shift is present in both the orientation analyzed, also if it is not of the same extent. The reported calculated results for the high concentration system refer to the face-to-back/back-to-back conformation. However, the two orientations clearly show the same qualitative behavior, pointing to a common underlying mechanism. The origin of the redox shift was demonstrated to be ascribable to the different polarizability of the environment that surrounds the QC in the two systems. In particular, in the low concentration system the environment is more polarizable than in the high concentration one, in which less water is present in the vicinity of the QC as an obvious consequence of the packing of the proteins on the electrode surface. This can be observed in Figure 5, where the distributions of the electrostatic potential exerted by the solvent (panels A and C) and by the rest of the perturbing environment (panels B and D) are reported for the low and high (face-to-face/back-to-back) concentration systems both in the reduced and in the oxidized ensemble. All the distributions are shifted toward smaller values upon oxidation, as a consequence of the positively charged quantum center in the oxidized state. However, the electrostatic potential due to water is noticeably more shifted in the low concentration system than in the high concentration one, thus implying that in the first one the solvent around the oxidized protein is more efficiently interacting with the protein, i.e. more polarized. On the contrary, the shift upon oxidation of the electrostatic potential due to the rest of the perturbing environment is almost the same in the two concentration states. Interestingly, also the difference in the extent of the shift between the two orientations can be explained with the same mechanism: the smaller shift obtained for the face-to-back system is indeed associated to a lesser displacement of solvent molecules than in the face-to-face/back-to-back one. This can be observed in Figure 6 where the normalized distribution of the number of water molecules within a radius of 1 nm from the heme group of the QC containing protein is reported, showing that more solvent molecules are close to the QC in the face-to-back system than in the face-to-face/back-to-back one.
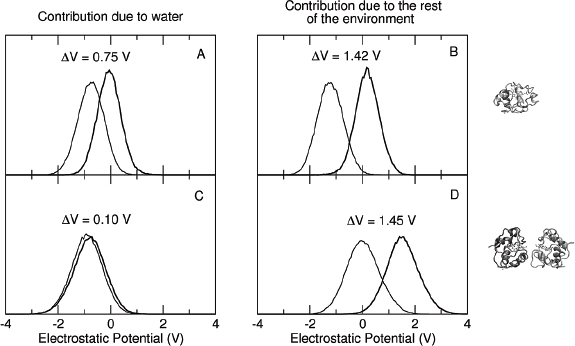
Figure 5 Normalized distribution of the electrostatic potential exerted on the active site by water (panels A and C) and from the rest of the perturbing environment (panels B and D) obtained from the simulations in the reduced (thick lines) and oxidized (thin lines) ensembles of the low (panels A and B) and high (panels C and D) concentration systems.
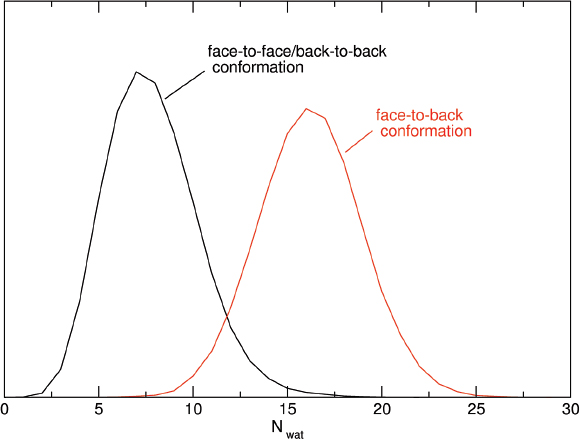
Figure 6 Normalized distribution of the number of water molecules within a radius of 1 nm from the heme group of the QC containing protein in the face-to-face/back-to-back simulation (black line) and in the face-to-back simulation (red line) Reprinted with permission from L. Zanetti-Polzi et al. J. Am. Chem. Soc., 136: 12929, 2014. Copyright 2014 American Chemical Society.
The reported results point to a key role of the solvent in determining the redox thermodynamics of Cyt c adsorbed on gold. It is worth to note that a significant role of water in determining the reduction potential of the same protein was also observed for Cyt c freely diffusing in solution, where the hydration state of the heme propionates was noted to be related to variations of the E0 value. [62]
More in general, the solvent fluctuations and the dynamic interaction between the solute and the solvent appear to be key factors in determining different properties of several biochemical systems. To gain insights into these kind of processes with theoretical/computational techniques, it is thus essential to maintain the configurational complexity of the environment, including water, and to achieve an extensive sampling of the dynamics of the system under investigation.
4 Conclusions
In this article, we have presented two comprehensive computational study of the interaction between proteins and GNPs. In the first presented case, we have introduced a detailed methodology involving multiple levels of theory, including docking by Brownian dynamics, classical atomistic MD and quantum mechanical DFT calculations which is able to provide molecular insights into the protein-GNP interactions that are not accessible from experiments alone. In particular, using our protein-surface docking strategy, we are able to discuss the nature of the interactions that guide the binding of a given protein to citrate-coated Au(111) and to observe the role of binding to citrate which represents the leading terms for the encounter complexes most compatible with experimental results. Moreover, the comparison between docking results obtained in various conditions (bare negative surface, citrate-covered surface) suggests a consistent accuracy of our developed parameters and a picture in which citrate molecules do remain on the surface of the NP, coexisting with the adsorbed protein. Analysis of the conformational changes of the protein data requires more extensive sampling methods and will be the subject of a forthcoming paper as well a direct comparison with experimental results. [46] In the second presented case, we have discussed a computational strategy based on a hybrid quantum mechanics/molecular mechanics methodology, the perturbed matrix method, which is able to accurately evaluate the redox potential of redox protein covalently bound to a gold electrode, reproducing the experimental conditions of an electrode with both low and high surface coverage. The computed values of the redox potentials are directly comparable with the experimental ones, as recently shown [32] and insights are gained into the molecular mechanism at the basis of the experimental shift observed between the low and high surface coverage case.
References
[1] M. Sarikaya, C. Tamerler, A. K. Jen, K. Schulten, F. Baneyx, Molecular Biomimetics: Nanotechnology through Biology. Nat. Mater., 2, 577-585 (2003).
[2] S. Linse, C. Cabaleiro-Lago, W.-F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford, K. A. Dawson, Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. USA, 104, 8691-8696 (2007).
[3] M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli, S. Laurent, Protein-Nanoparticle Interactions: Opportunities and Challenges. Chem. Rev., 111, 5610-5637 (2011).
[4] M.-E. Aubin-Tam, S. Hwang, and K Hamad-Schifferli, Site-Directed Nanoparticle labeling of Cytochrome C. Proc. Natl. Acad. Sci. U.S.A., 106, 4095-4100 (2009).
[5] G. Goobes, R. Goobes, O. Schueler-Furman, D. Baker, P. S. Stayton, Drobny, G. P. Folding of the C-Terminal Bacterial binding Domain in Statherin upon Adsorption onto Hydroxyapatite Crystals. Proc. Natl. Acad. Sci. U.S.A., 103, 16083-16088 (2006).
[6] J. J. Gray, The Interaction of Proteins with Solid Surfaces. Curr. Opin. Struct. Bio., 14, 110-115 (2004).
[7] J. H. Harding, D. M. Duffy, Sushko, L. Maria, P. Mark Rodger, D. Quigley, J. A. Elliott, Computational techniques at the organic-inorganic interface in biomineralization. Chem. Rev., 108, 4823-4854 (2008).
[8] R. A. Latour, Molecular Simulation of Protein-Surface Interactions: Benefits, Problems, Solutions, and Future Directions. Biointerphases, 3, FC2-FC12 (2008).
[9] O. Cohavi, S. Corni, F. De Rienzo, R. Di Felice, K. E. Gottschalk, M. Hoefling, D. Kokh, E. Molinari, G. Schreiber, A. Vaskevich, R. C. Wade, Protein-Surface Interactions: Challenging Experiments and Computations. J. Mol. Recognit., 23, 259-262 (2009).
[10] M. Bachmann, K. Goede, A. G. Beck-Sickinger, M. Grundmann, A. Irbäck, and W. Janke, Microscopic Mechanism of Specific Peptide Adhesion to Semiconductor Substrates. Angew. Chem., Int. Ed., 49, 9530-9533 (2010).
[11] R. Di Felice, S. Corni, Simulation of Peptide-Surface Recognition. J. Phys. Chem. Lett., 2, 1510-1519 (2011).
[12] K. Makrodimitris, D. L. Masica, E. T. Kim, J. J. Gray, Structure Prediction of Protein-Solid Surface Interactions Reveals a Molecular Recognition Motif of Statherin for Hydroxyapatite. J. Am. Chem. Soc., 129, 13713-13722 (2007).
[13] L. M. Ghiringhelli, B. Hess, N. F. A. van der Vegt, L. Delle Site, Competing Adsorption between Hydrated Peptides and Water onto Metal Surfaces: from Electronic to Conformational Properties. J. Am. Chem. Soc., 130, 13460-13464 (2008).
[14] G. Hong, H. Heinz, R. R. R. Naik, B. L. Farmer, R. Pachter, Toward Understanding Amino Acid Adsorption at Metallic Interfaces: a Density Functional Theory Study. ACS Appl. Mater. Interfaces, 1, 388-392 (2009).
[15] A. Vila Verde, J. M. Acres, J. K. Maranas, Investigating the Specificity of Peptide Adsorption on Gold Using Molecular Dynamics Simulations. Biomacromolecules, 10, 2118-2128 (2009).
[16] A. Vila Verde, P. J. Beltramo, J. K. Maranas, Adsorption of Homopolypeptides on Gold Investigated Using Atomistic Molecular Dynamics. Langmuir, 27, 10, 5918-5926 (2011).
[17] R. Coppage, J. M. Slocik, B. D. Briggs, A. I. Frenkel, H. Heinz, R. R. Naik, M. R. Knecht, Crystallographic Recognition Controls Peptide Binding for Bio-Based Nanomaterials. J. Am. Chem. Soc., 133, 12346-12349 (2011).
[18] A. Calzolari, G. Cicero, C. Cavazzoni, R. D. Felice, A. Catellani, S. Corni, Hydroxyl-Rich β-Sheet Adhesion to the Gold Surface in Water by First-Principle Simulations. J. Am. Chem. Soc., 132, 4790-4795 (2010).
[19] J. Yu, M. L. Becker, G. Carri, The Influence of Amino Acid Sequence and Functionality on the Binding Process of Peptides onto Gold Surfaces. Langmuir, 28, 1408-1417 (2012).
[20] L. Ruan, H. Ramezani-Dakhel, C. Y. Chiu, E. Zhu, Y. Li, H. H. Y. Heinz, Tailoring molecular specificity toward a crystal facet: a lesson from biorecognition toward Pt(111). Nano Lett., 13, 840-846 (2013).
[21] M. Hoefling, F. Iori, S. Corni, K. E. Gottschalk, The Conformations of Amino Acids on a Gold(111) Surface. ChemPhysChem, 11, 1763-1767 (2010).
[22] M. Hoefling, Iori F.; S. Corni, K. E. Gottschalk, Interaction of Amino Acids with the Au(111) Surface: Adsorption Free Energies from Molecular Dynamics Simulations. Langmuir, 26, 8347-8351 (2010).
[23] M. Hoefling, S. Monti, S. Corni, K. E. Gottschalk, Interaction of β-Sheet Folds with a Gold Surface. PLoS One, 6, e20925 (2012).
[24] D. Toroz, S. Corni, Peptide Synthesis of Gold Nanoparticles: the Early Steps of Gold Reduction Investigated by Density Functional Theory. Nano Lett., 11, 1313-1318 (2011).
[25] L. B. Wright, P. M. Rodger, S. Corni, T. R. Walsh, GolP-CHARMM: First-Principles Based Force Fields for the Interaction of Proteins with Au(111) and Au(100) J. Chem. Theory Comput., 9, 1616-1630 (2013).
[26] L. B. Wright, P. M. Rodger, T. R. Walsh, S. Corni, First-Principles-Based Force Field for the Interaction of Proteins with Au(100)(5x1): An Extension of GolP-CHARMM J. Phys. Chem. C, 117, 24292-24306 (2013).
[27] G. Brancolini, D. B. Kokh, L. Calzolai, R. C. Wade, S. Corni, Docking of Ubiquitin to Gold Nanoparticles. ACS NANO, 6, 9863-9878 (2012).
[28] G. Brancolini, D. Toroz, S. Corni, Can small hydrophobic gold nanoparticles inhibit β2-Microglobulin fibrillation? Nanoscale, 6, 7903-7911 (2014).
[29] F. Iori, R. Di Felice, E. Molinari, S. Corni, GolP: An Atomistic Force-Field to Describe the Interaction of Proteins with Au(111) Surfaces in Water J. Comp. Chem., 30, 1465-1476 (2009).
[30] M. Aschi, R. Spezia, A. Di Nola, and A. Amadei, A first priciples method to model perturbed electronic wavefunctions: the effect of an external electric field. Chem. Phys. Lett., 344:374-380, 2001.
[31] D. B. Kokh, S. Corni, P. J. Winn, M. Hoefling, K. E. Gottschalk, R. C. Wade, ProMetCS: An Atomistic Force Field for Modeling Protein-Metal Surface Interactions in a Continuum Aquesous Solvent. J. Chem. Theory Comput., 6, 1753-1768 (2010).
[32] L. Zanetti-Polzi, I. Daidone, C. A. Bortolotti, and S. Corni, Surface packing determines the redox potential shift of cytochrome c adsorbed on gold. Journal of the American Chemical Society, 136:12929-12937, 2014.
[33] R. R. Gabdoulline, R. C. Wade, Simulation of the Diffusional Association of Barnase and Barstar. Biophys. J., 72, 1917-1929 (1997).
[34] www.h-its.org/mcm
[35] D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark, Berendsen, H. J. C, GROMACS: Fast, Flexible, and Free. J. Comp. Chem., 26, 1701-1718 (2005).
[36] C. Oostenbrink, A. Villa, A. E. Mark, W. F. van Gunsteren, A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comp. Chem., 25, 1656-1676 (2004).
[37] N. Spackova, I. Berger, M. Egli, J. Sponer, Molecular Dynamics of Hemiprotonated Intercalated Four-Stranded i-DNA: Stable Stable Trajectories on a Nanosecond Scale J. Am. Chem. Soc., 120, 6147-6151 (1998).
[38] L. B. Wright, M. P. Rodgera, T. R. Walsh, Aqueous citrate: a first-principles and force-field molecular dynamics study RSC Adv., 3, 16399-16409 (2013).
[39] J.-W. Park, J. S. Shumaker-Parry, Structural Study of Citrate Layers on Gold Nanoparticles: Role of Intermolecular Interactions in Stabilizing Nanoparticles. J. Am. Chem. Soc., 136, 1907-1921 (2014).
[40] J. Kunze, I. Burgess, R. Nichols, I. Buess-Herman, J. Lipkowski, Electrochemical Evaluation of Citrate Adsorption on Au(111) and the Stability of Citrate-Reduced Gold Colloids. J. Electroanal. Chem., 599, 147-159 (2007).
[41] http://biophysics.cs.vt.edu/H++
[42] J. L. Elechiguerra, J. Reyes-Gasga, M. J. Yacaman, The Role of Twinning in Shape Evolution of Anisotropic Noble Metal Nanostructures J. Mater. Chem., 16, 3906-3919 (2006).
[43] Y. Lin, G. Pan, G.-J. Su, X.-H. Fang, L.-J. Wan, C.-L. Bai, Study of Citrate Adsorbed on the Au(111) Surface by Scanning Probe Microscopy. Langmuir, 19, 10000-10003 (2003).
[44] http://projects.villa-bosch.de/mcmsoft/sda/6.00/
[45] R. R. Gabdoulline, R. C. Wade, Effective Charges for Macromolecules in Solvent. J. Phys. Chem., 100, 3868-3878 (1996).
[46] G. Brancolini, A. Corazza, M. Vuano, F. Fogolari, M. C. Mimmi, V. Bellotti, M. Stoppini, S. Corni, G. Esposito, to be submitted 2014
[47] L. Zanetti-Polzi, A. Amadei, M. Aschi, and I. Daidone, Insight into the ir-spectra/structure relationship in amyloid fibrils: a theoretical study on a prion peptide. J. Am. Chem. Soc., 133(30):11414-11417 (2011).
[48] A. Amadei, I. Daidone, and M. Aschi, A general theoretical model for electron transfer reactions in complex systems. Phys. Chem. Chem. Phys., 14:1360-13770 (2012).
[49] I. Daidone, A. Amadei, F. Zaccanti, M. Borsari, and C. A. Bortolotti, How the reorganization free energy affects the reduction potential of structurally homologous cytochromes. The Journal of Physical Chemistry Letters, 5(9):1534-1540 (2014).
[50] R. Spezia, M. Aschi, A. Di Nola, and A. Amadei, Extension of the perturbed matrix method: application to a water molecule. Chem. Phys. Lett., 365:450-456 (2002).
[51] A. Amadei, M. D’Abramo, C. Zazza, and M. Aschi, Electronic properties of formaldehyde: a theoretical study. Chem. Phys. Lett., 381:187-193 (2003).
[52] A. Amadei, F. Marinelli, M. D’Abramo, M. D’Alessandro, M. Anselmi, A. Di Nola, and M. Aschi, Theoretical modeling of vibro-electronic quantum states in complex molecular systems: solvated carbon monoxide, a test case. J. Chem. Phys., 122:124506 (2005).
[53] A. Amadei, M. D'Alessandro, M. D'Abramo, and M. Aschi, Theoretical characterization of electronic states in interacting chemical systems. J. Chem Phys., 130:08410-08415 (2009).
[54] J. Gao and D. G. Truhlar, Quantum mechanical methods for enzyme kinetics. Ann. Rev. Phys. Chem., 53:467-505 (2002).
[55] T. Vreven and K. Morokuma, Chapter 3 hybrid methods: ONIOM (QM:MM) and QM/MM. Ann. Rep. Comp. Chem., 2:35-51 (2006).
[56] H. M. Senn and W. Thiel, QM/MM studies of enzymes. Curr. Opin. Chem. Biol., 11:182-187 (2007).
[57] A. Amadei, M. D'Alessandro, and M. Aschi, Statistical mechanical modeling of chemical reactions in complex systems: the reaction free energy surface. J. Phys. Chem. B, 108:16250-16254 (2004).
[58] I. Muegge, P. X. Qi, A. J. Wand, Z. T. Chu, and A. Warshel, The reorganization energy of cytochrome c revisited. J. Phys. Chem. B, 101:825-836 (1997).
[59] L. B. Sagle, J. Zimmermann, S. Matsuda, P. E. Dawson, and F. E. Romesberg, Redox-coupled dynamics and folding in cytochrome c. J. Am. Chem. Soc., 1128(24):7909-7915 (2006).
[60] H. A. Heering, F. G. M. Wiertz, C. Dekker, and S. deVries, Direct immobilization of native yeast iso-1 cytochrome c on bare gold: fast electron relay to redox enzymes and zeptomole protein-film voltammetry. J. Am. Chem. Soc., 126:11103-11112 (2004).
[61] C. A. Bortolotti, G. Battistuzzi, M. Borsari, P. Facci, A. Ranieri, and M. Sola, The redox chemistry of the covalently immobilized native and low-pH forms of yeast iso-1-cytochrome c. J. Am. Chem. Soc., 128:5444-5451 (2006).
[62] C. A. Bortolotti, A. Amadei, M. Aschi, M. Borsari, S. Corni, M. Sola, and I. Daidone, The reversible opening of water channels in cytochrome c modulates the heme iron reduction potential. J. Am. Chem. Soc., 134:13670-13678 (2012).
Biographies

G. Brancolini received her Ph.D. in Physical Chemistry from University of Modena and Reggio Emilia, in cooperation with the University of Ulm, where she spent 10 months at the Theoretical Physics Department, supported by a Marie Curie Fellowhip. She was a post-doctoral fellow at Politecnico di Milano from 2002-2004 in the Department of Chemical Engineering. From 2004-2010 she was a senior post-doctoral fellow at National Research Council in Italy. Since 2010 Dr. Brancolini is a fixed term research scientist at the Center S3 of the Institute of Nanoscience of the National Research Council (CNR-NANO-S3) in Modena, Italy.

L. Zanetti-Polzi got a Ph.D. in Biophysics at the University of Rome “La Sapienza” (Rome, Italy, 2010) working in the “Theoretical and Computational Physical Chemistry” group of Prof. A. Di Nola. She got two post-doctoral fellows in the same group and she is currently a post-doc at the Center S3 of the Institute of Nanoscience of the National Research Council (CNR-NANO-S3) in Modena, Italy. Her research activity mainly focuses on the simulation of molecular and macromolecular systems with classical and combined quantum/classical methods.

S. Corni is a tenured research scientist at the Center S3 of the Institute of Nanoscience of the National Research Council (CNR-NANO-S3) in Modena, Italy. He got a Ph.D. in Chemistry from Scuola Normale Superiore (Pisa, Italy, 2003) working with Jacopo Tomasi. His research interests cover various branches of physical chemistry and nanoscience, such as the interaction of (bio-)molecules with inorganic materials, the optical properties of molecule-nanoparticle hybrids, and electron transfer in biomolecules.