Integrating DNA with Functional Nanomaterials
Received 10 May 2013; Accepted 22 May 2013; Publication 6 June 2013
Journal of Self-Assembly and Molecular Electronics, Vol. 1, 177–194.
doi: 10.13052/jsame2245-4551.122
Copyright © 2013 River Publishers. All rights reserved.
Shalom J. Wind1, Erika Penzo1, Matteo Palma1, Risheng Wang1, Teresa Fazio1, Danny Porath2, Dvir Rotem2, Gideon Livshits2 and Avigail Stern2
- 1Department of Applied Physics and Applied Mathematics Columbia University, New York, NY, USA
- 2Department of Chemistry Hebrew University of Jerusalem, Jerusalem, Israel
1. Introduction
DNA may be the most versatile molecule discovered to date. Beyond its well-known central role in genetics, DNA has the potential to be a remarkably useful technological material. It has been demonstrated as a scaffold for the assembly of organic and inorganic nanomaterials [1]; a vehicle for drug delivery [2]; a medium for computation [3]; and a possible wire for transporting electrical signals [4]. A key factor in exploiting DNA in these ways is the ability to integrate DNA with other materials. In this paper, we review two approaches to forming DNA complexes with functional nanomaterials: (1) linking DNA with single-wall carbon nanotubes (SWCNTs), which can then be used as nanoscale electrical contacts for probing electron transport in DNA; and (2) directed nanoassembly of Au nanoparticles using DNA/PNA (peptide nucleic acid) hybrid scaffolds.
2. DNA-SWCNT Junctions
This work is motivated by the original work of Guo et al. [5, 6], which demonstrated the efficacy of SWNT electrodes for the study of charge transport through individual molecules. SWNTs are nearly ideal for this purpose. They are outstanding one-dimensional conductors, they can be linked to organic molecules through straightforward carbon-carbon chemistries, and they are essentially the same size (diameter) as individual molecules, ensuring that only a single molecule is being probed in each experiment. This platform was first applied to the study of charge transport in DNA by the Nuckolls group at Columbia, where they demonstrated efficient transport through well-matched dsDNA strands connected to SWCNTs via an amine linkage, supporting the contention that dsDNA contacted in this way maintains its native conformation [7]. The single-molecule devices in this and the previous work were fabricated by a process in which a nanoscalegap in a SWCNT is formed by “cutting” the SWCNT through a lithographically-defined stencil using an O2 plasma. This approach has been quite successful, in terms of demonstrating electron transport in DNA and other molecules, however, it is extremely inefficient; only ∼3% or fewer of cut nanotubes result in reconnection with the DNA. The primary reason for this is the difficulty in precisely matching the size of the opening to the length of the dsDNA molecule.
In order to overcome this difficulty, we have taken a new approach in which the most challenging aspect of the process, namely, formation of the connection between the DNA molecule and the SWCNT electrodes, would be achieved in solution by chemical means. Once these hybrid structures are formed, they could be placed on a surface for electrical measurement, using either a shadow mask electrode in conjunction with a conductive AFM tip (a technique developed by the Porath lab) or using pre-patterned electrodes on the surface. The extended length of the SWCNT-dsDNA hybrids renders them far easier to contact than individual DNA molecules.
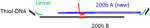
SWCNT-dsDNA hybrid complexes (Fig. 1) were created by reacting a water solution of SWCNT segments with amine functionalized dsDNA (26 base pairs mixed sequence, described below). Two reaction schemes were developed, one consisting of one step, the other consisting of two steps. The second scheme results in a higher yield of the desired CNT-dsDNA-CNT structure (Fig. 1).
Figure 1 Schematic of end-connected SWCNT-dsDNA Hybrid Structure.
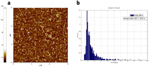
The starting material consisted of short SWCNT segments wrapped in single stranded DNA [DNA sequence: (GT)20] and dissolved in deionized water (concentration ∼40g/ml), obtained from M. Zheng at NIST. The solution is the result of a purification procedure based on size-exclusion chromatography (SEC) which sorts the CNT segments into fractions of uniform length [8]. The SWCNT segment length distribution was quantified by tapping mode AFM imaging (Fig. 2a) and software analysis (ImageJ, Fig. 2b). AFM samples were obtained by depositing 10l of CNT solution diluted 1:20 in deionized water, on a silicon dioxide substrate treated with oxygen plasma. The solution was dried in air and the samples were then washed by dipping them for 10 seconds in a solution of 50% DI water, 50% ethanol, then immersing them in a solution of 10% DI water, 90% ethanol for 50 minutes and finally letting them dry in air. The average length and standard deviation of the CNT segments in the starting solution were found to be 14893nm. Using this average length we estimated the molar concentration of CNT segments to be about 70 nM.
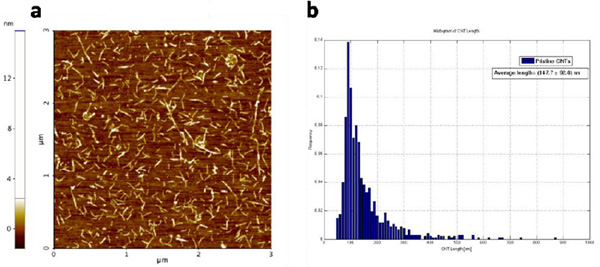
Figure 2 a) Tapping mode AFM image of the pristine DNA-wrapped SWCNT segments deionized water solution. (b) Histogram of the nanotube segments length distribution of the starting solution. The histogram was built by measuring the SWCNT length with ImageJ software. 1024 nanotube segments were measured, from different AFM images of different substrates on which the same solution was deposited. The average length and standard deviation of the SWCNT segments in the starting solution were found to be 148 93 nm
Two different schemes were followed to create the SWCNT-dsDNA hybrids. In the first scheme, a one-step reaction, the starting SWCNT solution was activated by mixing it, 1:1 by volume, with a solution consisting of 0.2 M MES buffer (pH 6), 4mM EDC and 10 mM sulfo-NHS. The SWCNTs sat in this solution for 30 min. at room temperature, during which the EDC and sulfo-NHS form an intermediate compound with the carboxyl groups on the CNT ends. Following activation, 2l of 0.5M amine-functionalized dsDNA (amine-26bp, described above) was added. The intermediate compound reacts with the amine groups on the DNA strands resulting in a covalent bond between the dsDNA and the SWCNT segments. This method for DNA attachment to acid-oxidized carbon nanotubes ends was previously reported by Weizmann et al. [10]. The concentration of amine-26bp during the reaction was 5 nM, corresponding to about one seventh of the nanotube concentration. The difference in concentration increases the probability that each DNA strand will react with two nanotube segments, one on each side, yielding the desired SWCNT-dsDNA hybrid structure. The mixture was left to react overnight at room temperature. Any unreacted DNA strands were removed by centrifugation in Millipore Amicon 100K tubes. During purification the buffer was exchanged to DPBS 1X.
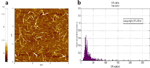
A histogram showing the length distribution of the resulting SWCNT-dsDNA hybrid structures (Fig. 3) was obtained by tapping mode AFM imaging and software analysis as explained above for the starting SWCNT solution. The average length and standard deviation of the CNT-dsDNA synthesized by this one step reaction were 325 283 nm.
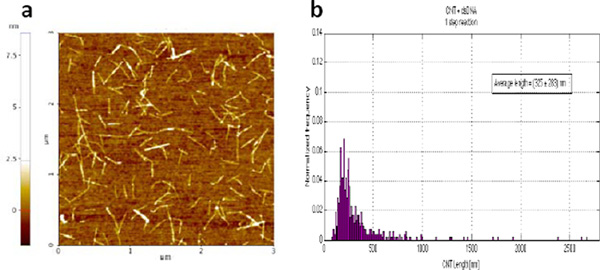
Figure 3 (a) Tapping mode AFM image of the SWCNT-dsDNA solution generated by the one-step reaction. (b) Histogram of the length distribution of the CNT-dsDNA hybrid structures obtained with the one-step reaction. The histogram was built by measuring the SWCNT length with ImageJ software. 525 nanotube segments were measured, from different AFM images of different substrates on which the same solution was deposited. The average length and standard deviation of the CNT segments in the starting solution were found to be 325 283 nm.
A second, two-step reaction scheme was also investigated. In this scheme, 20l of the starting SWCNT solution was mixed, 1:1 by volume, with a solution consisting of 0.2 M MES buffer, 4 mM EDC and 10 mM sulfo-NHS. This solution was let to activate for 30 min at room temperature before adding 20l of 0.5M amine-functionalized dsDNA (amine-26bp). The resulting concentration of amine-26bp was 167 nM, making it sufficiently likely that both ends of all SWCNT segments would be saturated. The mixture was left to react overnight at room temperature. Any unreacted DNA strands were removed by centrifugation in Millipore Amicon 100K tubes (the residual concentration of dsDNA after purification was estimated to be less than 0.5 nM). During purification the buffer was exchanged to DPBS 1X. 20l of the starting SWCNT solution was mixed, 1:1 by volume, to a solution consisting of 0.2 M MES buffer, 4 mM EDC and 10 mM sulfo-NHS. This solution was left to activate for 30 min. at room temperature before adding it to the same volume of the purified SWCNT-dsDNA. This second reaction resulted in the attachment of the activated SWCNT segments to the ones previously bound to the dsDNA.
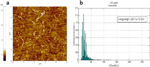
The size distribution of the resulting CNT-dsDNA hybrid structures is shown in Fig. 4. The average length and standard deviation of the CNT-dsDNA synthesized by this two steps reaction are 248113 nm.
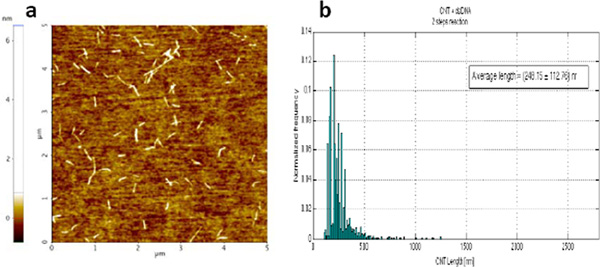
Figure 4 Tapping mode AFM image of the SWCNT-dsDNA solution generated by the two-step reaction. b) Histogram of the length distribution of the SWCNT-dsDNA hybrid structures obtained with the two-step reaction. The histogram was built by measuring the SWCNT length with ImageJ software. 1833 nanotube segments were measured, from different AFM images of different substrates on which the same solution was deposited. The average length and standard deviation of the CNT segments in the starting solution were found to be 248 113 nm.
Both the one-step and two-step synthesis schemes result in the formation of SWCNT-dsDNA hybrid structures, as demonstrated by the increase in the average length, shown in the two distributions. However, the two-step reaction produces a significantly narrower length distribution, which likely reflects a higher yield of the desired SWCNT-dsDNA hybrid structure. This is reasonable, since in the one-step scheme, a percentage of the SWCNT segments could be expected to be saturated with dsDNA on both ends, rendering them unable to link any further. The two-step scheme remedies this by introducing a new, unreacted population of activated SWCNTs which can bind with the saturated nanotubes.
Figure 5 shows a small area AFM scan of a dilute solution of SWCNT-dsDNA hybrids deposited on mica. Several different conformations can be observed, including both straight and kinked structures, which correspond to what would be expected for the SWCNT-dsDNA hybrids. Presumably, the kink could indicate the location of the dsDNA within the structure. We believe that these structures are well-suited for electrical characterization, the results of which will be reported separately.

Figure 5 AFM scan of SWCNT-dsDNA Hybrids on Mica Prior to Electrode Deposition
3. Nanoassembly using DNA-PNA Hybrids
Peptide nucleic acid (PNA) is an analogue of DNA with a backbone made from N-(2-aminoethyl glycine) units instead of DNA’s deoxyribose phosphate; the N-terminal of the PNA backbone corresponds to the 5’ end of the DNA backbone [11, 12]. The last decade has seen increased attention paid and progress made in developing PNA targeting reagents and methods. PNA has been explored for uses in drug development and diagnostics [13]. Moreover, PNA binding to DNA can affect gene expression, and offers increased chemical stability in the presence of other enzymes. For example, PNA bound to a restriction enzyme site inhibits site cleavage by that enzyme [14]. PNA has also been explored for use with DNA origami as part of DX (double crossover) molecules [15]. In this way, it has been incorporated into arrays of 2D nucleic acid tiles, a useful tool for self-assembling large-scale arrays on surfaces.
PNA can bind to DNA in several ways: duplex formation with ssDNA or RNA, triplex formation by Watson-Crick and Hoogsteen binding, and strand displacement for Watson-Crick binding of PNA base pairs to the DNA duplex [12]. PNA also forms double helical structures nearly identical to DNA. For this reason, design of complementary PNA labels is highly sequence and site-specific. In this work, we employ strand displacement of part of one strand of the DNA duplex in order to bind a PNA-DNA chimera to dsDNA [16]. Complementary base-pair binding can be used for DNA labeling; an identical backbone is not necessary for hybridization of labels by base pairs. Whereas DNA has a negatively charged backbone, the backbone of PNA is neutral. PNA-DNA duplexes are more stable at higher temperatures than DNA-DNA duplexes; the melting temperature of a PNA-DNA duplex increases in melting temperature by 1°C per base pair over a DNA-DNA duplex in the presence of 100mM NaCl [17]. Strand displacement/invasion of DNA by PNA takes advantage of the breathing modes inherent in heated DNA. When DNA is heated, the individual strands come apart slightly, allowing PNA to preferentially bind to the complementary base pairs. If PNA is introduced into the solution at this time, as the solution cools, the base pairs of the PNA strand will preferentially pair with complementary base pairs on the DNA, as there is less electrostatic repulsion as in DNA-DNA binding. The PNA forms a triplex, creating a loop in the DNA.
Stadler et al. proposed that naturally-occurring lambda DNA could be used as a scaffold for assembly of functional nanomaterials [16]. Toward that end, they demonstratedsolution-based binding of Au nanoparticle to synthetic double-stranded DNA via duplex invasion of a PNA-DNA chimera [16]. We sought to build upon this method by optimizing the binding conditions to control the placement of nanoparticles on a surface, with an eye toward ultimately using naturally-occurring -DNA in order to extend the length scales across which DNA-modulated architectures could be self-assembled. It is possible to tune the PNA sequence and incubation time and temperature in order to invade specific sites on -DNA, which measures 16 m in length [18, 19]. This could enable site-selective placement of nanoscale architectures over length scales far exceeding the dimensions of DNA origami. One caveat is that the properties of differently-synthesized PNA greatly affect binding specificity and yield. Put simply, every different PNA designed may require a different binding process; there is no one-size-fits-all method of determining binding conditions for every PNA molecule.
To test the binding of PNA to lambda-DNA fragments, binding conditions were adapted from Chan et al and Zohar et al. [18, 19]. The PNA was a bis- with sequence: N-dig-OO-Lys-Lys-TCC TTC TC-OOO-JTJ TTJ JT-Lys-OO-Lys-O-COOH [18, 19]. The “J” base pair is pseudoisocytosine, which can replace cytosine in the sequence and reduces the binding sensitivity to ionic strength. This sequence preferentially invades DNA using triplex invasion [20]. Triplex invasion complexes are very stable at high temperatures. Lambda DNA (NEB, 48kbp) was digested with NcoI and XbaI for 1.5hrs at 37°C in NEB4 buffer, yielding a 607bp fragment. PNA (Biosynthesis, Inc., Lewisville, TX) was incubated with the restricted lambda for 25 hours at 37°C in the presence of 2mM NaCl, thenallowed to cool to room temperature on the benchtop. The gel was stained with SYBR green, an ultrasensitive dye, for imaging via 4% polyacrylamide gel-shift assay.
We also examined binding of PNA to a 45bp synthesized lambda DNA fragment [21]. The DNA sequence used was: 5’-GCA ACA GTG GCATGC ACC GAG AAG GAC GTT TGT AAT GTC CGCTCC-3’, which is a fragment of lambda DNA from base pairs 24342–24386, with melting temperature 70.5°C.
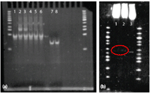
PNA was mixed with single-stranded complementary and non-complementary strands in the following molar ratios to the single strands: 10:1, 5:1, 1:1, 0.5:1, and 0.1:1. Although we followed the protocols for binding PNA to DNA from Chan et al.[18] and Zohar et al.[19], The oligos were incubated with PNA for a shorter time because of their reduced size as compared to full lambda DNA. Briefly, PNA was added to DNA with 100 mM NaCl in 1X Tris-EDTA bufer and incubated in a water bath at 63°C for 1.5hrs. then allowed cool to room temperature. A 4 polyacrylamide gel was run at 90V for 2 hrs. We confirmed the binding of PNA to 45 base-pair dsDNA duplexes. In panel (a) of Figure 6, gel shifts can clearly be seen with PNA hybridized to DNA at molar ratios of 10:1 and 5:1. Gel-shift experiments with larger lambda fragments from restriction digests were less consistent. Experiments involving PNA bound to a restriction-digested enzyme were inconsistent, but it is thought that a gel-shift occurred with a 607bp fragment, as in Figure 6, panel (b).
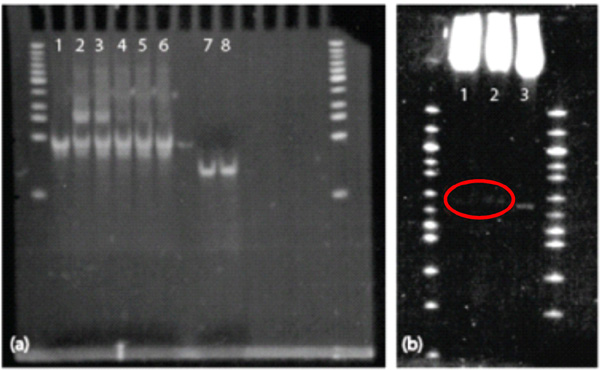
Figure 6 Gel-Shift Assays. PNA binds well to the 45bp dsDNA fragments at higher molarratios, but is barely visible (and not supershifted) on the larger restriction-enzyme-digested portion. (a) PNA gel-shift assay on 45bp dsDNA duplexes. Column 1 is 45bp dsDNA; 2 shows a gel shift due to PNA bound to DNA at a 10:1 molar ratio; 3 shows a gel shift due to PNA binding DNA at a 5:1 molar ratio. Columns 4, 5, and 6 show no gel shift; PNA was incubated with DNA at molar ratios of 1:1, 0.5:1, and 0.1:1, respectively. Column 7 shows 45bp ssDNA, and Column 8 is ssDNA incubated with 0.1:1 PNA:DNA (shows nobinding). (b) PNA gel-shift assay on 607bp fragment. Columns 1 and 2 shows gel shiftson 607bp lambda + PNA + an anti-digoxigenin Fab fragment and 607bp lambda + PNA, respectively. Column 3 shows the 607bp fragment alone.
Hybridization of PNA to DNA was performed following the methods of Stadler et al.[16]. In this method, 200 base-pair fragments of synthesized dsDNA are incubated with 25 base-pair long single-stranded (ss) PNA-DNA chimeras at 50 °C overnight, allowing the single-stranded PNA-DNA chimera to preferentially invade the charged DNA helix. Although this labeling was been done in solution, we sought to perform directed self-assembly of DNA structures on lithographically-patterned surfaces, increasing nanopattern resolution and allowing self-assembly of functional nanomaterials on these surfaces. Placement of DNA molecules on a surface can be directed by binding them between patterned nanodots, which are functionalized with complementary oligonucleotides. This assembly scheme is illustrated in Figure 7.
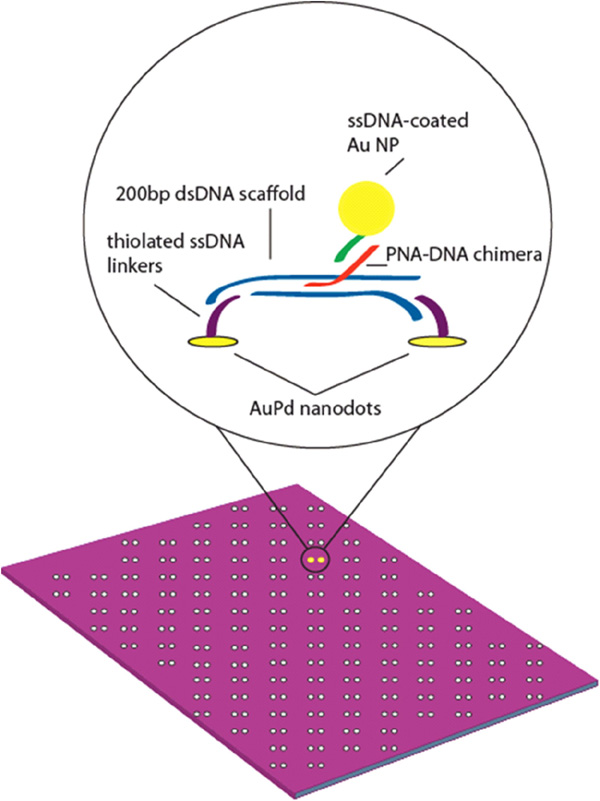
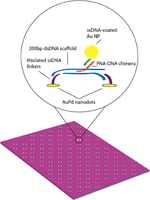
Figure 7 Schematic of Au NPs bound to PNA-DNA chimera – labeled dsDNA scaffold on AuPd nanodot-patterned surface.
The nanodots anchors were formed by nanoimprint lithography (NIL) and selective pattern transfer [22] on oxidized Si substrates. The Si NIL template was patterned by electron beam lithography using hydrogen silsesquioxane (HSQ) as a resist [23]. After annealing in air for densification, the HSQ itself served as the three-dimensional relief structure for NIL. Thermal NIL of poly-methylmethacrylate (PMMA) and subsequent pattern transfer processes yielded approximately spherical nanodots, with uniform diameter ∼7 – 8 nm. The nanodots were patterned in arrays of dimers with an inter-dot spacing of 60nm.
The nanodots were functionalized with ssDNA using a thiol linker [24]. The following sequence was used: 5-ThioMC6-D / TTT TTTTTTTTTTTT TAA CCT AAC CTT CAT; the 5' thiolated end bound to the AuPd dots. Prior to hybridization to thessDNA linkers, duplex invasion of a PNA-DNA chimera was used to functionalize a 200bp fragment of dsDNA[c16]. Briefly, PNA was incubated with DNA in a 3X molar excess at 50 °C overnight in 10 mM phosphate buffer with 100mM NaCl. After overnight incubation, the resulting solution was left to cool to room temperature. Scaffolds were incubated with an equal molar concentration of linker arm ssDNA for several hours at room temperature.
The scaffold consisted of a 200bp dsDNA duplex with different length linker arms. The sequences of the two halves of the zigzag structure (shown in Fig. 8) were: 5-ACG TAC CAA ATA CGT CGA TTG GCT ACG TAA TAA CAA TTT CTA TTG GTT CCG CAA GCT GGC CCT CAC TTC AAC GCA TTG TTA TTA ATC TTC CAA TGG GCC ACC TAC CGT AGA CAC GGA CTC TCT ACG CGT TAT GCC TCA GCA TAT TGT TAT TAC TGC GGG ACA TAC GAT AGA GCT TTG CTA AAA TAA GTC CCT GCC TT-3 and 5-ACG TAC CAA ATA CGT CGA TTG GCT ACG TAA TAA CAA TTT CTA TTG GTG GAA AGG CAG GGA CTT ATT TTA GCA AAG CTC TAT CGT ATG TCC CGC AGT AAT AAC AAT ATG CTG AGG CAT AAC GCG TAG AGA GTC CGT GTC TAC GGT AGG TGG CCC ATT GGA AGA TTA ATA ACA ATG CGT TGA AGT GAG GGC CAG CTT GCG GA-3 with the PNA-binding sequence in bold. The sequence of the linker arms was: 5-ACG TAT TTG GTA CGT (T)n ATG AAG GTT AGG TTA-3, where poly-T chains of n 18, 36, and 54 were inserted into the linker to extend their length and allow for more room for binding to occur.
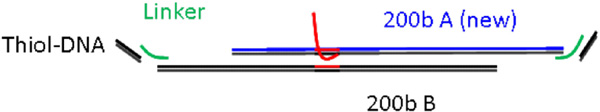
Figure 8 Schematic of the Zigzag DNA Scaffold, Including 3' Sticky Ends. The Linker Length was Varied to Optimize the Binding Percentage.
The sequence of the PNA-DNA chimera, which bound to the bold section of the DNA, was: N-TAA TAA CAA T-Linker-CAC ATC TCT TCT GAA -3, with the PNA sequence in bold. After overnight incubation of ssDNA on nanodots, substrates were removed from hybridization buffer, dip-rinsed in a beaker of PBS, and partially dried with a laboratory wipe until only a thin film of PBS buffer remained on the substrate. The substrates were then incubated with 20L of 100–200n DNA-PNA scaffold solution and incubated motionless at room temperature for 6–8 hours. Following incubation of the PNA-containing 200bp scaffold, a 795nM solution of 10 nm gold nanoparticles coated with ssDNA complementary to the ssDNA portion of the PNA-DNA chimera was added. The sample was incubated overnight at room temperature to allow the nanoparticles to bind to the scaffold via the PNA-DNA chimera.
The binding yield was determined by SEM analysis. Approximately 500 or more sites for each sample were measured. Nanoparticles within a 30nm radius of the AuPd nanodots were deemed to be tethered by one end of the dsDNA duplex, while nanoparticles between the dots counted as trimers tethered by both ends of the dsDNA duplex. To determine the degree of non-specific nanoparticle binding to the surface, control experiments were performed with no PNA-DNA chimera attached to the scaffolds.
Linker arm lengths were varied in order to determine the optimal length for binding, and how much extra length was necessary to accommodate the nanoparticle binding to the PNA-DNA chimera and subsequent surface deposition. Poly-thymine linkers with lengths of 18, 36, and 54 base pairs were used. These made total scaffold lengths of 236, 272, and 290 base pairs, respectively. Table 5.1 illustrates results of experiments with varying linker lengths.
Stadler et al. were able to achieve a binding yield of ∼26% in solution [16]. In analyzing surface binding to nanodots with 60nm spacing, we found that the best results came from a linker length involving the 36-T linker. In this case, 8.5% of dot-pairs had a gold nanoparticle in between them. An additional 13.4% of pairs had an AuNP nearby, indicating monovalent DNA-PNA scaffold tethering to a single AuPd nanodot. This contributes to a total of ∼22% of dot pairs which have a nanoparticle self-assembled to the scaffold containing the DNA-PNA chimera, as shown in Fig. 9. This surface-based self-assembly result corresponds to nearly 85% of the solution yield. By contrast, only ∼10% of dot pairs using scaffolds with 18-T and 54-T linkers displayed trimers or single-tethered nanoparticles. This may be explained by the 18-T linker being too short, while the 54-T linker is too long for efficient binding. Control experiments using a scaffold with no attached PNA-DNA chimera show ∼6% of dot pairs with a nanoparticle nearby due to nonspecific Au nanoparticle binding or AuPd nanopattern defects.

Figure 9 Directed assembly of PNA-labeled DNA scaffolds on AuPd dot pairs. Positions of trimers are highlighted by white ellipses.
Nanofabricated templates offer a starting point for directed self-assembly of double-stranded DNA arrays. As demonstrated here and elsewhere, site-specific labeling of dsDNA at tunable sites is possible using PNA, LNA, and nick-translation in solution [16, 18, 19]. In this work, annealing PNA to DNA was done using only short (∼45–600bp) double-stranded DNA fragments. Moreover, we have demonstrated surface-based self-assembly of gold nanoparticles onto a double-stranded DNA scaffold via PNA-DNA and DNA base-pair interactions. However, PNA invasion is extremely sensitive to conditions such as temperature, time, any linkers present (e.g. digoxigenin or other haptens) and base-pair sequence. Therefore, it remains difficult to massively tune hybridization conditions for PNA on lambda DNA. For DNA fragments up to 200 base pairs, PNA invasion works in solution for up to 26% of the sample [16]. Therefore, a rate of ∼22% for surface-based assembly of nanoparticles in between and around AuPd nanodots constitutes 85% of solution-based binding efficiency. Labeling this DNA via PNA increases the resolution of these methods by enabling the site-specific placement of functional nano-objects (e.g. Au nanoparticles, quantum dots, nanowires, etc.) on surfaces. This has positive implications for self-assembly of larger, more complicated structures on surfaces.
4. Conclusions
In this work, we have demonstrated two approaches to integrating DNA with functional nanomaterials. One approach results in a hybrid nanostructure that has the potential to be further integrated into an active nanoelectronic device (i.e., where the DNA serves as the active element). The other approach demonstrates a new way to look at DNA as a scaffold for directed assembly. Together, both demonstrate the versatility of DNA as a technological material that can be exploited for many future applications.
Acknowledgements
We thank Oleg Gang, Andrea Stadler and Peter Sun at Brookhaven National Lab for their help, guidance and material support with PNA/DNA hybridization. We gratefully acknowledge financial support from the US-Israel Binational Science Foundation under Award SIBSF2006422 and the Office of Naval Research under Award N00014-09-1-1117. Additionalsupport from the Nanoscale Science and Engineering Initiative of the National Science Foundation under NSF Award CHE-0641523 and from the New York State Office of Science, Foundationunder NSF Award CHE-0641523 and from the New York State Office of Science, Technology, and Academic Research (NYSTAR) is also gratefully acknowledged.
References
[1] F. A. Aldaye, A. L. Palmer, and H. F. Sleiman, Assembling Materials with DNA as the Guide, Science, 321(5897): 1795–1799 (2008).
[2] J. Fu and H. Yan, Controlled drug release by a nanorobot, Nat Biotech, 30(5): 407–408 (2012).
[3] L. M. Adleman, Computing with DNA, Sci Am, 279(2): 54–61 (1998).
[4] J. C. Genereux and J.K. Barton, Mechanisms for DNA Charge Transport, Chem Rev, 110(3):1642–1662 (2010).
[5] X. Guo, J. P. Small, J. E. Klare, Y. Wang, M. S. Purewal, I. W. Tam, B. H. Hong, R. Caldwell, L. Huang, S. O'Brien, J. Yan, R. Breslow, S. J. Wind, J. Hone, P. Kim, and C. Nuckolls, Covalently bridging-gaps in single-walled carbon nanotubes with conducting molecules, Science, 311(5759): 356–359 (2006.).
[6] X. F. Guo, A. Whalley, J. E. Klare, L. M. Huang, S. O'Brien, M. Steigerwald, and C. Nuckolls, Single-molecule devices as scaffolding for multicomponent nanostructure assembly, Nano Lett,. 7(5):1119–1122 (2007).
[7] X. F. Guo, A. A. Gorodetsky, J. Hone, J. K. Barton, and C. Nuckolls, Conductivity of a single DNA duplex bridging a carbon nanotube gap. Nature Nanotechnology, 3(3): 163–167 (2008).
[8] X. Y. Huang, R. S. McLean, and M. Zheng, High-resolution length sorting and purification of DNA-wrapped carbon nanotubes by size-exclusion chromatography, Anal Chem, 77(19): 6225–6228 (2005).
[9] DNA strands were purchased from Syntezza.
[10] Y. Weizmann, D. M. Chenoweth, and T. M. Swager, Addressable Terminally Linked DNA-CNT Nanowires, J Am Chem Soc, 132(40):14009–14011 (2010).
[11] T. Bentin and P. E. Nielsen, In vitro transcription of a torsionally constrained template, Nucleic Acids Res, 30(3): 803–809 (2002).
[12] P. E. Nielsen, M. Egholm, and O. Buchardt, Peptide Nucleic-Acid (PNA) – a DNA Mimic with a Peptide Backbone, Bioconjugate Chem, 1994. 5(1): p. 3–7.
[13] P. E. Nielsen, A new molecule of life? Peptide nucleic acid, a synthetic hybrid of protein and DNA, could form the basis of a new class of drugs- and of artificial life unlike anything found in nature, Sci Am, 299(6): 64–71 (2008).
[14] P. E. Nielsen, M. Egholm, R. H. Berg, and O. Buchardt, Sequence Specific-Inhibition of DNA Restriction Enzyme Cleavage by PNA, Nucleic Acids Res, 1993. 21(2): 197–200.
[15] P. S. Lukeman, A. C. Mittal, and N. C. Seeman, Two dimensional PNA/DNA arrays: estimating the helicity of unusual nucleic acid polymers, Chem Commun, (15): 1694–1695 (2004).
[16] A. L. Stadler, D.Z. Sun, M. M. Maye, D. van der Lelie, and O. Gang, Site-Selective Binding of Nanoparticles to Double-Stranded DNA via Peptide Nucleic Acid “Invasion”, ACS Nano, 5(4): 2467–2474 (2011).
[17] Applied biosystems – support. https://www2.appliedbiosystems.com/support/seqguide.cfm?
[18] E. Y. Chan, N. M. Goncalves, R. A. Haeusler, A. J. Hatch, J. W. Larson, A. M. Maletta, G. R. Yantz, E.D. Carstea, M. Fuchs, G.G. Wong, S.R. Gullans, and R. Gilmanshin, DNA mapping using microfluidic stretching and single-molecule detection of fluorescent site-specific tags, Genome Res, 14(6): 1137–1146 (2004).
[19] H. Zohar, C. L. Hetherington, C. Bustamante, and S. J. Muller, Peptide Nucleic Acids as Tools for Single-Molecule Sequence Detection and Manipulation, Nano Lett, 10(11): 4697–4701 (2010).
[20] P. E. Nielsen, Peptide nucleic acids : methods and protocols. Methods in molecular biology., Totowa, N. J.: Humana Press. 274 p (2002).
[21] Integrated DNA Technologies, Coralville, IA
[22] M. Schvartzman and S.J. Wind, Robust Pattern Transfer of Nanoimprinted Features for Sub-5-nm Fabrication. Nano Lett, 9(10): 3629–3634 (2009).
[23] H. Namatsu, Y. Takahashi, K. Yamazaki, T. Yamaguchi, M. Nagase, and K. Kurihara, Three-dimensional siloxane resist for the formation of nanopatterns with minimum linewidth fluctuations, J Vac Sci Technol B, 16(1): 69–76 (1998).
[24] M. Palma, J. J. Abramson, A. A. Gorodetsky, E. Penzo, R. L. Gonzalez, M. P. Sheetz, C. Nuckolls, J. Hone, and S. J. Wind, Selective Biomolecular Nanoarrays for Parallel Single-Molecule Investigations, J Am Chem Soc, 133(20): 7656–7659 (2011).
Biographies

Shalom J. Wind received his Ph.D. in Physics from Yale University in 1987. Following his doctoral studies, he worked at IBM’s Thomas J. Watson Research Center, focusing primarily on nanoelectronic devices. He moved to the Department of Applied Physics and Applied Mathematics at Columbia University in 2003. Dr. Wind’s present research focuses on molecular-scale fabrication and the interface between biological and technological materials and systems.

Erika Penzo was born in Schio, Italy, in 1983. She got her BSc and MSc in Physics Engineering from Politecnico di Milano, Italy. She is currently a PhD candidate at Columbia University (New York) in the Applied Physics department. Her research focuses on nanofabrication, self-assembly and DNA nanotechnology.

Matteo Palma received his Ph.D. in Physical Chemistry from University Louis Pasteur in 2007. He was a post-doctoral fellow at Columbia University from 2008–2012 in the Departments of Mechanical Engineering and Applied Physics and Applied Mathematics. Dr. Palma will take up a position as a lecturer at Queen Mary College, University of London, beginning in September 2013. Dr. Palma’s research interests include the controlled self-assembly of functional nanostructures on surfaces with true nanoscale resolution, with a particular focus on supramolecular interactions to drive the self-organization of nano-moieties on (nanopatterned) substrates.